3-hydroxy-3-methylglutaryl-CoA lyase
| Hydroxymethylglutaryl-CoA lyase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
HMG-CoA lyase dimer, Human | |||||||||
| Identifiers | |||||||||
| EC number | 4.1.3.4 | ||||||||
| CAS number | 9030-83-5 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / EGO | ||||||||
| |||||||||

| 3-hydroxymethyl-3-methylglutaryl-Coenzyme A lyase (hydroxymethylglutaricaciduria) | |
|---|---|
| Identifiers | |
| Symbol | HMGCL |
| Entrez | 3155 |
| HUGO | 5005 |
| OMIM | 246450 |
| RefSeq | NM_000191 |
| UniProt | P35914 |
| Other data | |
| EC number | 4.1.3.4 |
| Locus | Chr. 1 p36.1-p35 |
3-hydroxy-3-methylglutaryl-CoA lyase (or HMG-CoA lyase) is an enzyme that in human is encoded by the HMGCL gene located on chromosome 1. It is a key enzyme in ketogenesis (ketone body formation).
Structure
The HMGCL gene encodes a 34.5-kDa protein that is localized to the mitochondrion and peroxisome.[1] Multible isoforms of the proteins are known due to alternative splicing. The major isoform (isoform 1) is most highly expressed in the liver [2] whereas isoform 2 is found in energy-demanding tissues including the brain, heart, and skeletal muscle.[3]
Structure of the HMGCL protein has been resolved by X-ray crystallography at 2.1-Å resolution, and reveals that the protein may function as a dimer. Substrate access to the active site of the HMGCL enzyme involves substrate binding across a cavity located at the C-terminal end of a beta barrel structure.[4] In addition, the lysine 48 residue which is mutated in patients with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency is also found to be necessary for substrate binding.[5]
Function
The HMGCL protein plays an essential role in breaking down dietary proteins and fats for energy. It catalyzes the reaction:
(S)-3-hydroxy-3-methylglutaryl-CoA = acetyl-CoA + acetoacetate.
and requires a divalent metal ion as co-factor.[6]
The enzyme is required for ketogenesis in the liver, and is also responsible for processing the amino acid leucine inside the mitochondrion
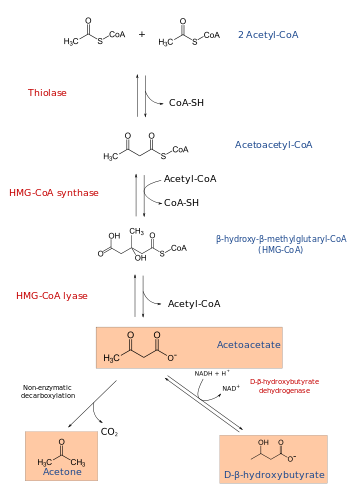
Clinical Significance
Mutations in the HMGCL gene cause 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMGCLD), a rare autosomal recessive inborn error of metabolism characterized by disruption of ketogenesis and L-leucine catabolism. To-date more than 30 different mutations including missense mutations of different residues have been associated with patients with HMGCLD in diverse families and ethnicities.[7] HMGCLD typically presents in the first year of the patient's life after a fasting period. Clinical acute symptoms include vomiting, seizures, metabolic acidosis, hypoketotic hypoglycemia, and lethargy.[8]
Interactions
HMGCL interacts with itself to form homodimers and homotetramers. It is also shown in yeast two-hybrid experiments to interact with DNAJA1.
References
- ↑ Ashmarina LI, Robert MF, Elsliger MA, Mitchell GA (1996). "Characterization of the hydroxymethylglutaryl-CoA lyase precursor, a protein targeted to peroxisomes and mitochondria". Biochem. J. 315 ( Pt 1): 71–5. PMC 1217198
 . PMID 8670134.
. PMID 8670134. - ↑ Ashmarina LI, Robert MF, Elsliger MA, Mitchell GA (1996). "Characterization of the hydroxymethylglutaryl-CoA lyase precursor, a protein targeted to peroxisomes and mitochondria". Biochem. J. 315 ( Pt 1): 71–5. PMC 1217198. PMID 8670134.
- ↑ Puisac B, Ramos M, Arnedo M, Menao S, Gil-Rodríguez MC, Teresa-Rodrigo ME, Pié A, de Karam JC, Wesselink JJ, Giménez I, Ramos FJ, Casals N, Gómez-Puertas P, Hegardt FG, Pié J (2012). "Characterization of splice variants of the genes encoding human mitochondrial HMG-CoA lyase and HMG-CoA synthase, the main enzymes of the ketogenesis pathway". Mol. Biol. Rep. 39 (4): 4777–85. doi:10.1007/s11033-011-1270-8. PMID 21952825.
- ↑ Fu Z, Runquist JA, Montgomery C, Miziorko HM, Kim JJ (2010). "Functional insights into human HMG-CoA lyase from structures of Acyl-CoA-containing ternary complexes". J. Biol. Chem. 285 (34): 26341–9. doi:10.1074/jbc.M110.139931. PMC 2924059. PMID 20558737.
- ↑ Carrasco P, Menao S, López-Viñas E, Santpere G, Clotet J, Sierra AY, Gratacós E, Puisac B, Gómez-Puertas P, Hegardt FG, Pie J, Casals N (2007). "C-terminal end and aminoacid Lys48 in HMG-CoA lyase are involved in substrate binding and enzyme activity". Mol. Genet. Metab. 91 (2): 120–7. doi:10.1016/j.ymgme.2007.03.007. PMID 17459752.
- ↑ Tuinstra RL, Miziorko HM (2003). "Investigation of conserved acidic residues in 3-hydroxy-3-methylglutaryl-CoA lyase: implications for human disease and for functional roles in a family of related proteins". J. Biol. Chem. 278 (39): 37092–8. doi:10.1074/jbc.M304472200. PMID 12874287.
- ↑ Menao S, López-Viñas E, Mir C, Puisac B, Gratacós E, Arnedo M, Carrasco P, Moreno S, Ramos M, Gil MC, Pié A, Ribes A, Pérez-Cerda C, Ugarte M, Clayton PT, Korman SH, Serra D, Asins G, Ramos FJ, Gómez-Puertas P, Hegardt FG, Casals N, Pié J (2009). "Ten novel HMGCL mutations in 24 patients of different origin with 3-hydroxy-3-methyl-glutaric aciduria". Human Mutation. 30 (3): E520–9. doi:10.1002/humu.20966. PMID 19177531.
- ↑ http://www.omim.org/entry/246450[]
External links
- 3-hydroxy-3-methylglutaryl-coenzyme A lyase at the US National Library of Medicine Medical Subject Headings (MeSH)
