Pulmonary hypertension
| Pulmonary hypertension | |
|---|---|
.jpg) | |
| Micrograph showing pulmonary hypertensive arteriopathy with marked thickening of the tunica intima and tunica media. | |
| Classification and external resources | |
| Specialty | Pulmonology, cardiology |
| ICD-10 | I27.0, I27.2 |
| ICD-9-CM | 416.0, 416.8 |
| DiseasesDB | 10998 |
| MedlinePlus | 000112 |
| eMedicine | radio/583 med/1962 Secondary pulmonary hypertension Pediatric primary pulmonary hypertension Persistent newborn pulmonary hypertension |
| Patient UK | Pulmonary hypertension |
| MeSH | D006976 |
Pulmonary hypertension (PH or PHTN) is an increase of blood pressure in the pulmonary artery, pulmonary vein, or pulmonary capillaries, together known as the lung vasculature, leading to shortness of breath, dizziness, fainting, leg swelling and other symptoms.[1] Pulmonary hypertension can be a severe disease with a markedly decreased exercise tolerance. It was first identified by Ernst von Romberg in 1891.[2] According to the most recent classification, it can be one of six different types.[3]
Signs and symptoms
.jpg)
The signs/symptoms of pulmonary hypertension include the following:[4][5][6]
- Shortness of breath
- Fatigue
- Chest pain
- Palpitations (heartbeat rate increased)
- Pain (right side of the abdomen)
- Poor appetite
- Lightheadedness
- Fainting or syncope
- Swelling (legs/ankles)
- Cyanosis
Less common signs/symptoms include non-productive cough and exercise-induced nausea and vomiting.[5] Coughing up of blood may occur in some patients, particularly those with specific subtypes of pulmonary hypertension such as heritable pulmonary arterial hypertension, Eisenmenger syndrome and chronic thromboembolic pulmonary hypertension.[7] Pulmonary venous hypertension typically presents with shortness of breath while lying flat or sleeping (orthopnea or paroxysmal nocturnal dyspnea), while pulmonary arterial hypertension (PAH) typically does not.[8]
Other typical signs of pulmonary hypertension include an accentuated pulmonary component of the second heart sound, a right ventricular third heart sound, and parasternal heave indicating a hypertrophied right atrium. Signs of systemic congestion resulting from right-sided heart failure include jugular venous distension, ascites, and hepatojugular reflux.[5][6][9] Evidence of tricuspid insufficiency and pulmonic regurgitation is also sought and, if present, is consistent with the presence of pulmonary hypertension.[5][6][10]
Causes
In terms of causes and classification,[11] a 1973 meeting organized by the World Health Organization was the first to attempt classification of pulmonary hypertension. A distinction was made between primary and secondary PH, and primary PH was divided in the "arterial plexiform", "veno-occlusive" and "thromboembolic" forms.[12] A second conference in 1998 at Évian-les-Bains also addressed the causes of secondary PH (i.e. those due to other medical conditions),[13] and in 2008, the 4th World Symposium on Pulmonary Arterial Hypertension was convened in Dana Point to modify the classification based on new understandings of disease mechanisms. The revised system developed by this group provides the current framework for understanding pulmonary hypertension.[3] The system includes several improvements over the former 2004 Venice Classification system.
The Dana Point 2008 Updated Clinical Classification system can be summarized as follows:[3]
- WHO Group I - Pulmonary arterial hypertension (PAH)
- Idiopathic PAH
- Heritable
- BMPR2
- ALK1, endoglin (with or without hereditary hemorrhagic telangiectasia)
- Unknown
- Drug- and toxin-induced
- Associated with
- Persistent pulmonary hypertension of the newborn
- WHO Group I' - Pulmonary veno-occlusive disease (PVOD) and/or pulmonary capillary hemangiomatosis (PCH)
- WHO Group II - Pulmonary hypertension owing to left heart disease
- WHO Group III - Pulmonary hypertension owing to lung disease and/or hypoxia
- Chronic obstructive pulmonary disease (COPD)
- Interstitial lung disease
- Other pulmonary diseases with mixed restrictive and obstructive pattern
- Sleep-disordered breathing
- Alveolar hypoventilation disorders
- Chronic exposure to high altitude
- Developmental abnormalities
- WHO Group IV - Chronic thromboembolic pulmonary hypertension (CTEPH)
- WHO Group V - Pulmonary hypertension with unclear multifactorial mechanisms
- Hematologic diseases: myeloproliferative disease, removal of the spleen
- Systemic diseases: sarcoidosis, pulmonary Langerhans cell histiocytosis: lymphangioleiomyomatosis, neurofibromatosis, vasculitis
- Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid diseases
- Others: tumoral obstruction, fibrosing mediastinitis, chronic kidney failure on dialysis
Pathogenesis
The pathogenesis of pulmonary arterial hypertension (WHO Group I) involves the narrowing of blood vessels connected to and within the lungs. This makes it harder for the heart to pump blood through the lungs, much as it is harder to make water flow through a narrow pipe as opposed to a wide one. Over time, the affected blood vessels become stiffer and thicker, in a process known as fibrosis. This further increases the blood pressure within the lungs and impairs their blood flow. In common with other types of pulmonary hypertension, the increased workload of the heart causes hypertrophy of the right ventricle, making the heart less able to pump blood through the lungs, ultimately causing right heart failure. The right ventricle is normally part of a low pressure system, with pressures that are lower than those that the left ventricle normally encounters. As such, the right ventricle cannot cope as well with higher pressures, and although hypertrophy of the heart muscle helps initially, it ultimately leads to a situation where the right ventricular muscle cannot get enough oxygen to meet its needs and right heart failure follows. As the blood flowing through the lungs decreases, the left side of the heart receives less blood. This blood may also carry less oxygen than normal. Therefore, it becomes harder and harder for the left side of the heart to pump to supply sufficient oxygen to the rest of the body, especially during physical activity.[14][15]
Pathogenesis in pulmonary hypertension owing to left heart disease (WHO Group II) is completely different in that constriction or damage to the pulmonary blood vessels is not the issue. Instead, the left heart fails to pump blood efficiently, leading to pooling of blood in the lungs and back pressure within the pulmonary system. This causes pulmonary edema and pleural effusions.[16]
In hypoxic pulmonary hypertension (WHO Group III), the low levels of oxygen are thought to cause narrowing of the pulmonary arteries. This phenomenon is called hypoxic pulmonary vasoconstriction and it is initially a protective response designed to stop too much blood flowing to areas of the lung that are damaged and do not contain oxygen. When the damage is widespread and prolonged, this hypoxia-mediated vasoconstriction occurs across a large portion of the pulmonary vascular bed.[17][18]
In chronic thromboembolic pulmonary hypertension (WHO Group IV), the blood vessels are blocked or narrowed with recurrent blood clots, and these clots can lead to release of substances that cause the blood vessels to constrict. This combination of blocked or narrowed vessels and vasoconstriction once again increases the resistance to blood flow and so the pressure within the system rises.[19]
Molecular pathology
The molecular mechanism of pulmonary arterial hypertension (PAH) is not known yet, but it is believed that the endothelial dysfunction results in a decrease in the synthesis of endothelium-derived vasodilators such as nitric oxide and prostacyclin.[20] Moreover, there is a stimulation of the synthesis of vasoconstrictors such as thromboxane and vascular endothelial growth factor (VEGF). These results in a severe vasoconstriction and smooth muscle and adventitial hypertrophy characteristic of patients with PAH.[21]
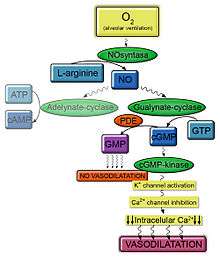
In normal conditions, the nitric oxide synthase produces nitric oxide from L-arginine in presence of oxygen.[22] Adenylate-cyclase and gualynate-cyclase are activated in presence of nitric oxide and these enzymes produce cAMP and cGMP respectively. The cGMP is produced by a type of guanylate cyclase (which is a kind of pyrophosphate-liase cyclase): the soluble guanylate cyclase (or sGC), catalyzes the formation of cGMP from GTP. sGC is a heterodimer made up of one α subunit and one β sub-unit in each chain. It also contains a prosthetic heme group, required for NO binding. The union of NO and sGC produces a conformational enzyme change that stimulates cGMP production.[23]
In the vascular endothelium, cGMP activates cGMP kinase or PKG (protein kinase G), which is an enzyme that belongs to a type of serine/threonine - specific protein kinase.[24] PKG is a dimer composed of two similar polypeptides chains that share a common molecular structure. Each subunit contains a catalytic domain and regulatory domain.[25] GMP-kinase activates potassium channels and subsequently the inhibition of calcium channels. Thus, this process leads to a reduction of intracellular calcium and finally a vasodilation.[26]
Phosphodiesterase type V (PDE5), which is abundant in the pulmonary tissue, is a metalohydrolase that hydrolyzes the cyclic bond of cGMP in the presence of divalent cations (Zn2+). Actually, Zn2+ union is necessary for PDE5 activity. In the N-terminal region (regulatory domain) of PDE5 there is an aminoacid sequence (residues 142-526) that joins cGMP. This sequence of PDE5 is divided in two domains; GAF-A and GAF-B; but only GAF-A has the necessary affinity to bind cGMP. This union increases the catalytic activity and it is stabilized by a close serine phosphorylation (performed by a kinase). Consequently, the concentration of cGMP decreases and the vasodilation is stopped.[23] Patients with PAH produce less NO and others vasodilators and produce more vasoconstrictors. Consequently, this molecular pathway doesn’t work properly and it results in a constant vasoconstriction. For this reason, NO and PDE5 inhibitors such as tadalafil or sildenafil are possible therapies.[26]
Diagnosis
In terms of the diagnosis of pulmonary hypertension, dictates it can be of five major types, a series of tests must be performed to distinguish pulmonary arterial hypertension from venous, hypoxic, thromboembolic, or miscellaneous varieties. Further procedures are required to confirm the presence of pulmonary hypertension and exclude other possible diagnoses. These generally include pulmonary function tests; blood tests to exclude HIV, autoimmune diseases, and liver disease; electrocardiography (ECG); arterial blood gas measurements; X-rays of the chest (followed by high-resolution CT scanning if interstitial lung disease is suspected); and ventilation-perfusion or V/Q scanning to exclude chronic thromboembolic pulmonary hypertension.[27][28] Clinical improvement is often measured by a "six-minute walk test", i.e. the distance a patient can walk in six minutes. Stability and improvement in this measurement correlate with better survival.[29]

Diagnosis of PAH requires the presence of pulmonary hypertension. Although pulmonary arterial pressure can be estimated on the basis of echocardiography,[30] pressure measurements with a Swan-Ganz catheter through the right side of the heart provide the most definite assessment.[31] Diagnosis of PAH requires right-sided cardiac catheterization; a Swan-Ganz catheter can also measure the cardiac output, which is far more important in measuring disease severity than the pulmonary arterial pressure.[31] Normal pulmonary arterial pressure in a person living at sea level has a mean value of 8–20 mm Hg (1066–2666 Pa) at rest. Pulmonary hypertension is present when mean pulmonary artery pressure exceeds 25 mm Hg (3300 Pa) at rest.[32]
Physical examination
A physical examination is performed to look for typical signs of pulmonary hypertension (described above).,[28] and a detailed family history is established to determine whether the disease might be heritable.[33][34][35] A history of exposure to drugs such as benfluorex (a fenfluramine derivative), dasatinib, cocaine, methamphetamine, ethanol leading to cirrhosis, and tobacco leading to emphysema is considered significant.[6][36][37] Use of selective serotonin reuptake inhibitors during pregnancy (particularly late pregnancy) is associated with an increased risk of the baby developing persistent pulmonary hypertension of the newborn.[37]
Echocardiography
A meta-analysis of doppler echocardiography for predicting right heart catheterization reported a sensitivity and specificity of 88% and 56%, respectively.[38]
Treatment
Treatment of pulmonary hypertension is determined by whether the PH is arterial, venous, hypoxic, thromboembolic, or miscellaneous. The treatment is to optimize left ventricular function by the use of diuretics, digoxins, blood thinners, or to repair/replace the mitral valve or aortic valve.[39] Patients with left heart failure or hypoxemic lung diseases (groups II or III pulmonary hypertension) should not routinely be treated with vasoactive agents including prostanoids, phosphodiesterase inhibitors, or endothelin antagonists, as these are approved for the different condition called pulmonary arterial hypertension.[40] To make the distinction, doctors at a minimum will conduct cardiac catheterization of the right heart, echocardiography, chest CT, a six-minute walk test, and pulmonary function testing.[40] Using treatments for other kinds of pulmonary hypertension in patients with these conditions can harm the patient and wastes substantial medical resources.[40]
High dose calcium channel blockers are useful in only 5% of IPAH patients who are vasoreactive by Swan-Ganz catheter. Unfortunately, calcium channel blockers have been largely misused, being prescribed to many patients with non-vasoreactive PAH, leading to excess morbidity and mortality.[10] The criteria for vasoreactivity have changed. Only those patients whose mean pulmonary artery pressure falls by more than 10 mm Hg to less than 40 mm Hg with an unchanged or increased cardiac output when challenged with adenosine, epoprostenol, or nitric oxide are considered vasoreactive.[41] Of these, only half of the patients are responsive to calcium channel blockers in the long term.[42]
A number of agents have recently been introduced for primary and secondary PAH. The trials supporting the use of these agents have been relatively small, and the only measure consistently used to compare their effectivity is the "6 minute walk test". Many have no data on mortality benefit or time to progression.[43]
Vasoactive substances
Many pathways are involved in the abnormal proliferation and contraction of the smooth muscle cells of the pulmonary arteries in patients with pulmonary arterial hypertension. Three of these pathways are important since they have been targeted with drugs — endothelin receptor antagonists, phosphodiesterase type 5 (PDE-5) inhibitors, and prostacyclin derivatives.[44]
Prostaglandins
Prostacyclin (prostaglandin I2) is commonly considered the most effective treatment for PAH. Epoprostenol (synthetic prostacyclin) is given via continuous infusion that requires a semi-permanent central venous catheter. This delivery system can cause sepsis and thrombosis. Prostacyclin is unstable, and therefore has to be kept on ice during administration. Since it has a half-life of 3 to 5 minutes, the infusion has to be continuous, and interruption can be fatal.[45] Other prostanoids have therefore been developed. Treprostinil can be given intravenously or subcutaneously, but the subcutaneous form can be very painful. An increased risk of sepsis with intravenous Remodulin has been reported by the CDC. Iloprost is also used in Europe intravenously and has a longer half life. Iloprost was the only inhaled form of prostacyclin approved for use in the US and Europe, until the inhaled form of treprostinil was approved by the FDA in July 2009.
Endothelin receptor antagonists
The dual (ETA and ETB) endothelin receptor antagonist bosentan was approved in 2001. Sitaxentan (Thelin) was approved for use in Canada, Australia, and the European Union,[46] but not in the United States. In 2010, Pfizer withdrew Thelin worldwide because of fatal liver complications. A similar drug, ambrisentan is marketed as Letairis in the U.S. by Gilead Sciences.[47]
Phosphodiesterase type 5 inhibitors
The U.S. FDA approved sildenafil, a selective inhibitor of cGMP specific phosphodiesterase type 5 (PDE5), for the treatment of PAH in 2005. It is marketed for PAH as Revatio. In 2009, they also approved tadalafil, another PDE5 inhibitor, marketed under the name Adcirca.[48] PDE5 inhibitors are believed to increase pulmonary artery vasodilation, and inhibit vascular remodeling, thus lowering pulmonary arterial pressure and pulmonary vascular resistance.[49]
Tadalafil is taken orally, as well as sildenafil, and it is rapidly absorbed (serum levels are detectable at 20 minutes). The T1/2 (biological half-life) hovers around 17.5 hours in healthy subjects.[50] Moreover, if we consider pharmacoeconomic implications, patients that take tadalafil would pay two-thirds of the cost of sildenafil therapy.[51] However, there are some adverse effects of this drug such as headache, diarrhea, nausea, back pain, dyspepsia, flushing and myalgia.[52]
Activators of soluble guanylate cyclase
Soluble guanylate cyclase (sGC) is the intracellular receptor for NO. As of April 2009, the sGC activators cinaciguat and riociguat were undergoing clinical trials for the treatment of PAH.[53]
Surgical
Atrial septostomy is a surgical procedure that creates a communication between the right and left atria. It relieves pressure on the right side of the heart, but at the cost of lower oxygen levels in blood (hypoxia). Lung transplantation cures pulmonary arterial hypertension, but leaves the patient with the complications of transplantation, and a post-surgical median survival of just over five years.[54]
Pulmonary thromboendarterectomy (PTE) is a surgical procedure that is used for chronic thromboembolic pulmonary hypertension. It is the surgical removal of an organized thrombus (clot) along with the lining of the pulmonary artery; it is a very difficult, major procedure that is currently performed in a few select centers.[55]
Monitoring
Established clinical practice guidelines dictate the frequency of pulmonary nodule evaluation and surveillance,[40][56] patients are normally monitored through commonly available tests such as:
- Pulse oximetry
- Arterial blood gas tests
- Chest X-rays
- Serial ECG tests
- Serial echocardiography
- Spirometry or more advanced lung function studies
Prognosis
.jpg)
The prognosis of pulmonary arterial hypertension (WHO Group I) has an untreated median survival of 2–3 years from time of diagnosis, with the cause of death usually being right ventricular failure (cor pulmonale).[57] A recent outcome study of those patients who had started treatment with bosentan (Tracleer) showed that 89% patients were alive at 2 years.[58] With new therapies, survival rates are increasing. For 2,635 patients enrolled in The Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Disease Management (REVEAL Registry) from March 2006 to December 2009, 1-, 3-, 5-, and 7-year survival rates were 85%, 68%, 57%, and 49%, respectively. For patients with idiopathic/familial PAH, survival rates were 91%, 74%, 65%, and 59%.[59] Levels of mortality are very high in pregnant women with severe pulmonary arterial hypertension (WHO Group I). Pregnancy is sometimes described as contraindicated in these women.[60][61][62]
Epidemiology
The epidemiology of IPAH is about 125-150 deaths per year in the U.S., and worldwide the incidence is similar to the U.S. at 4 cases per million. However, in parts of Europe (France) indications are 6 cases per million of IPAH. Females have a higher incidence rate than males (2-9:1).[63]
Other forms of PH are far more common. In systemic scleroderma, the incidence has been estimated to be 8 to 12% of all patients;[64] in rheumatoid arthritis it is rare.[65] However, in systemic lupus erythematosus it is 4 to 14%,[66] and in sickle cell disease, it ranges from 20 to 40%.[67] Up to 4% of people who suffer a pulmonary embolism go on to develop chronic thromboembolic disease including pulmonary hypertension.[68] A small percentage of patients with COPD develop pulmonary hypertension with no other disease to explain the high pressure.[69] On the other hand, obesity-hypoventilation syndrome is very commonly associated with right heart failure due to pulmonary hypertension.[70]
Notable cases
- Elaine Kaufman, American restaurateur[71]
- Priya Balachandran, TV producer [72]
- Ina Balin, American Broadway and tv actress[73]
- Chloe Temtchine, American singer-songwriter[74][75][76]
- Natalie Cole[77]
See also
References
- ↑ "Pulmonary hypertension: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. Retrieved 2015-12-30.
- ↑ von Romberg, Ernst (1891–1892). "Über Sklerose der Lungenarterie". Dtsch Arch Klin Med (in German). 48: 197–206.
- 1 2 3 Simonneau G, Robbins I, Beghetti M, et al. (30 June 2009). "Updated Clinical classification of pulmonary hypertension". J. Am. Coll. Cardiol. 54 (1 Suppl S): S43–S54. doi:10.1016/j.jacc.2009.04.012. PMID 19555858.
- ↑ "What Are the Signs and Symptoms of Pulmonary Hypertension? - NHLBI, NIH". www.nhlbi.nih.gov. Retrieved 2015-12-30.
- 1 2 3 4 Galie N, Humbert M, Vachiery JL, et al. (1 January 2016). "2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT)". European Heart Journal. 37 (1): 67–119. doi:10.1093/eurheartj/ehv317. ISSN 1522-9645. PMID 26320113.
- 1 2 3 4 McLaughlin VV, Archer SL, Badesch DB, et al. (28 April 2009). "ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association". Journal of the American College of Cardiology. 53 (17): 1573–1619. doi:10.1016/j.jacc.2009.01.004. ISSN 1558-3597. PMID 19389575.
- ↑ Diller, Gerhard-Paul; Gatzoulis, Michael A. (27 February 2007). "Pulmonary vascular disease in adults with congenital heart disease". Circulation. 115 (8): 1039–1050. doi:10.1161/CIRCULATIONAHA.105.592386. ISSN 1524-4539. PMID 17325254.
- ↑ Fang JC, DeMarco T, Givertz MM, et al. (1 September 2012). "World Health Organization Pulmonary Hypertension group 2: pulmonary hypertension due to left heart disease in the adult--a summary statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation". The Journal of Heart and Lung Transplantation: The Official Publication of the International Society for Heart Transplantation. 31 (9): 913–933. doi:10.1016/j.healun.2012.06.002. ISSN 1557-3117. PMID 22884380.
- ↑ Yusuf, Salim; Cairns, John; Camm, John; Fallen, Ernest L.; Gersh, Bernard J. (2011-09-07). Evidence-Based Cardiology. John Wiley & Sons. p. 70.3(figure). ISBN 9781444359459.
- 1 2 "Primary Pulmonary Hypertension Clinical Presentation: History, Physical Examination, Complications". emedicine.medscape.com. Retrieved 2015-12-30.
- ↑ "National Guideline Clearinghouse | ACR Appropriateness Criteria® pulmonary hypertension.". www.guideline.gov. Retrieved 2015-12-31.
- ↑ Hatano S, Strasser R (1975). Primary pulmonary hypertension. Geneva: World Health Organization.
- ↑ Rich S, Rubin LJ, Abenhail L, et al. (1998). Executive summary from the World Symposium on Primary Pulmonary Hypertension (Evian, France, September 6–10, 1998). Geneva: The World Health Organization. Archived from the original on April 8, 2002.
- ↑ Yuan, Jason X.-J.; Rubin, Lewis J. (2005-02-08). "Pathogenesis of Pulmonary Arterial Hypertension The Need for Multiple Hits". Circulation. 111 (5): 534–538. doi:10.1161/01.CIR.0000156326.48823.55. ISSN 0009-7322. PMID 15699271.
- ↑ Tuder, Rubin M.; Marecki, John C.; Richter, Amy; Fijalkowska, Iwona; Flores, Sonia (2007-03-01). "Pathology of Pulmonary Hypertension". Clinics in chest medicine. 28 (1): 23–vii. doi:10.1016/j.ccm.2006.11.010. ISSN 0272-5231. PMC 1924722
 . PMID 17338926.
. PMID 17338926. - ↑ Guazzi, Marco; Galiè, Nazzareno (2012-12-01). "Pulmonary hypertension in left heart disease". European Respiratory Review. 21 (126): 338–346. doi:10.1183/09059180.00004612. ISSN 0905-9180. PMID 23204122.
- ↑ Sommer, N.; Dietrich, A.; Schermuly, R. T.; Ghofrani, H. A.; Gudermann, T.; Schulz, R.; Seeger, W.; Grimminger, F.; Weissmann, N. (2008-12-01). "Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms". European Respiratory Journal. 32 (6): 1639–1651. doi:10.1183/09031936.00013908. ISSN 0903-1936. PMID 19043010.
- ↑ Stenmark, Kurt R.; Fagan, Karen A.; Frid, Maria G. (2006-09-29). "Hypoxia-Induced Pulmonary Vascular Remodeling Cellular and Molecular Mechanisms". Circulation Research. 99 (7): 675–691. doi:10.1161/01.RES.0000243584.45145.3f. ISSN 0009-7330. PMID 17008597.
- ↑ McNeil, Keith; Dunning, John (2007-09-01). "Chronic thromboembolic pulmonary hypertension (CTEPH)". Heart. 93 (9): 1152–1158. doi:10.1136/hrt.2004.053603. ISSN 1355-6037. PMC 1955041. PMID 17699182.
- ↑ Budhiraja, Rohit; Tuder, Rubin M.; Hassoun, Paul M. (2004-01-20). "Endothelial Dysfunction in Pulmonary Hypertension". Circulation. 109 (2): 159–165. doi:10.1161/01.CIR.0000102381.57477.50. ISSN 0009-7322. PMID 14734504.
- ↑ Budhiraja R, Tuder RM, Hassoun. PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165.
- ↑ Förstermann, Ulrich; Münzel, Thomas (2006-04-04). "Endothelial Nitric Oxide Synthase in Vascular Disease From Marvel to Menace". Circulation. 113 (13): 1708–1714. doi:10.1161/CIRCULATIONAHA.105.602532. ISSN 0009-7322. PMID 16585403.
- 1 2 Fosfodiesterasas del AMPc y del GMPc en el cerebro: Expresión en procesos neuroinflamatorios y neurodegenerativos. URL: http://www.tesisenred.net/bitstream/handle/10803/891/03.ERI_METODOS.pdf?sequence=4. Viewed 3 November 2012.|Text in Spanish
- ↑ Golan, David E. (2008-01-01). Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy. Lippincott Williams & Wilkins. p. 370. ISBN 9780781783552.
- ↑ Taylor, C. M. (2012-12-02). Intracellular Messengers. Newnes. ISBN 9780080966946.
- 1 2 Ghofrani HA, Pepke-Zaba J, Barbera JA; et al. (2004). "Nitric oxide pathway and phosphodiesterase inhibitors in pulmonary arterial hypertension". J Am Coll Cardiol. 43: 68S–72S.
- ↑ Members, Authors/Task Force; Galiè, Nazzareno; Hoeper, Marius M.; Humbert, Marc; Torbicki, Adam; Vachiery, Jean-Luc; Barbera, Joan Albert; Beghetti, Maurice; Corris, Paul (2009-10-01). "Guidelines for the diagnosis and treatment of pulmonary hypertension". European Heart Journal. 30 (20): 2493–2537. doi:10.1093/eurheartj/ehp297. ISSN 0195-668X. PMID 19713419.
- 1 2 "How Is Pulmonary Hypertension Diagnosed? - NHLBI, NIH". www.nhlbi.nih.gov. Retrieved 2015-12-30.
- ↑ "Guidelines:Six-minute Walk Test" (PDF). American Thoracic Society. 2002. Retrieved 31 December 2015.
- ↑ Bossone, Eduardo (2011). "Echocardiography in Pulmonary Arterial Hypertension:From Diagnosis to Prognosis" (PDF). Journal of the American Society of Cardiology. Retrieved 31 December 2015.
- 1 2 "Swan-Ganz - right heart catheterization: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. Retrieved 2015-12-30.
- ↑ Badesch, DB; Champion, HC; Sanchez, MA; Hoeper, MM; Loyd, JE; Manes, A; McGoon, M; Naeije, R; Olschewski, H; Oudiz, RJ; Torbicki, A (Jun 30, 2009). "Diagnosis and assessment of pulmonary arterial hypertension.". Journal of the American College of Cardiology. 54 (1 Suppl): S55–66. doi:10.1016/j.jacc.2009.04.011. PMID 19555859.
- ↑ "Pulmonary arterial hypertension". Genetics Home Reference. 2015-12-28. Retrieved 2015-12-30.
- ↑ Austin, Eric D.; Loyd, James E.; Phillips, John A. (1993-01-01). Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C., eds. Heritable Pulmonary Arterial Hypertension. Seattle (WA): University of Washington, Seattle. PMID 20301658.|Online 2015
- ↑ Hoeper MM, Bogaard HJ, Condliffe R, et al. (24 December 2013). "Definitions and diagnosis of pulmonary hypertension". Journal of the American College of Cardiology. 62 (25 Suppl): D42–50. doi:10.1016/j.jacc.2013.10.032. ISSN 1558-3597. PMID 24355641.
- ↑ Klepper, Michael J.; Cobert, Barton (2010-10-25). Drug Safety Data: How to Analyze, Summarize and Interpret to Determine Risk. Jones & Bartlett Learning. p. 86. ISBN 9780763769123.
- 1 2 Simonneau G, Gatzoulis MA, Adatia I, et al. (24 December 2013). "Updated clinical classification of pulmonary hypertension". Journal of the American College of Cardiology. 62 (25 Suppl): D34–41. doi:10.1016/j.jacc.2013.10.029. ISSN 1558-3597. PMID 24355639.
- ↑ Taleb M, Khuder S, Tinkel J, Khouri SJ (2013). "The diagnostic accuracy of Doppler echocardiography in assessment of pulmonary artery systolic pressure: a meta-analysis.". Echocardiography. 30 (3): 258–65. doi:10.1111/echo.12061. PMID 23227919.
- ↑ "How Is Pulmonary Hypertension Treated? - NHLBI, NIH". www.nhlbi.nih.gov. Retrieved 2015-12-30.
- 1 2 3 4 American College of Chest Physicians; American Thoracic Society (September 2013), "Five Things Physicians and Patients Should Question", Choosing Wisely: an initiative of the ABIM Foundation, American College of Chest Physicians and American Thoracic Society, retrieved 6 January 2013, which cites
- McLaughlin, V. V.; Archer, S. L.; Badesch, D. B.; Barst, R. J.; Farber, H. W.; Lindner, J. R.; Mathier, M. A.; McGoon, M. D.; Park, M. H.; Rosenson, R. S.; Rubin, L. J.; Tapson, V. F.; Varga, J.; Harrington, R. A.; Anderson, J. L.; Bates, E. R.; Bridges, C. R.; Eisenberg, M. J.; Ferrari, V. A.; Grines, C. L.; Hlatky, M. A.; Jacobs, A. K.; Kaul, S.; Lichtenberg, R. C.; Lindner, J. R.; Moliterno, D. J.; Mukherjee, D.; Pohost, G. M.; Rosenson, R. S.; Schofield, R. S. (2009). "ACCF/AHA 2009 Expert Consensus Document on Pulmonary Hypertension: A Report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: Developed in Collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association". Circulation. 119 (16): 2250–2294. doi:10.1161/CIRCULATIONAHA.109.192230. PMID 19332472.*Galie, N.; Hoeper, M. M.; Humbert, M.; Torbicki, A.; Vachiery, J. -L.; Barbera, J. A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J. S.; Gomez-Sanchez, M. A.; Jondeau, G.; Klepetko, W.; Opitz, C.; Peacock, A.; Rubin, L.; Zellweger, M.; Simonneau, G.; Vahanian, A.; Auricchio, A.; Bax, J.; Ceconi, C.; Dean, V.; Filippatos, G.; Funck-Brentano, C.; Hobbs, R.; Kearney, P.; McDonagh, T.; McGregor, K.; Popescu, B. A.; et al. (2009). "Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT)". European Heart Journal. 30 (20): 2493–2537. doi:10.1093/eurheartj/ehp297. PMID 19713419.*Hoeper, M. M.; Barberà, J. A.; Channick, R. N.; Hassoun, P. M.; Lang, I. M.; Manes, A.; Martinez, F. J.; Naeije, R.; Olschewski, H.; Pepke-Zaba, J.; Redfield, M. M.; Robbins, I. M.; Souza, R. R.; Torbicki, A.; McGoon, M. (2009). "Diagnosis, Assessment, and Treatment of Non-Pulmonary Arterial Hypertension Pulmonary Hypertension". Journal of the American College of Cardiology. 54 (1 Suppl): S85–S96. doi:10.1016/j.jacc.2009.04.008. PMID 19555862.
- ↑ Barst RJ, McGoon M, Torbicki A, et al. (June 2004). "Diagnosis and differential assessment of pulmonary arterial hypertension". J. Am. Coll. Cardiol. 43 (12 Suppl S): 40S–47S. doi:10.1016/j.jacc.2004.02.032. PMID 15194177.
- ↑ Sitbon O, Humbert M, Jaïs X, et al. (June 2005). "Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension". Circulation. 111 (23): 3105–11. doi:10.1161/CIRCULATIONAHA.104.488486. PMID 15939821.
- ↑ Torres F (2007). "Systematic review of randomised, double-blind clinical trials of oral agents conducted in patients with pulmonary arterial hypertension". Int. J. Clin. Pract. 61 (10): 1756–65. doi:10.1111/j.1742-1241.2007.01545.x. PMID 17877662.
- ↑ Raja, Shahzad G.; Raja, Shahbaz M. (2011-11-01). "Treating pulmonary arterial hypertension: current treatments and future prospects". Therapeutic Advances in Chronic Disease. 2 (6): 359–370. doi:10.1177/2040622311420773. ISSN 2040-6223. PMC 3513893. PMID 23251761.
- ↑ "Treatment of pulmonary arterial hypertension: The role of prostacyclin and prostaglandin analogs - Respiratory Medicine". www.resmedjournal.com. Retrieved 2015-12-30.
- ↑ "UPDATE 1-Encysive gets Canadian approval for hypertension drug". Reuters. 2008-05-30. Retrieved 2007-07-08.
- ↑ "U.S. Food and Drug Administration Approves Gilead's Letairis Treatment of Pulmonary Arterial Hypertension" (Press release). Gilead Sciences. 2007-06-15. Retrieved 2007-06-16.
- ↑ "FDA approves Adcirca (tadalafil) tablets for pulmonary arterial hypertension" (Press release). 2009-05-26. Retrieved 2010-12-06.
- ↑ Duarte, Julio D; Hanson, Rebekah L; Machado, Roberto F (2013-05-01). "Pharmacologic treatments for pulmonary hypertension: exploring pharmacogenomics". Future cardiology. 9 (3): 335–349. doi:10.2217/fca.13.6. ISSN 1479-6678. PMC 3864092. PMID 23668740.
- ↑ Forgue ST, Patterson BE, Bedding AW; et al. (2005). "Tadalafil pharmacokinetics in healthy subjects". Br J Clin Pharmacol. 61: 280–288. doi:10.1111/j.1365-2125.2005.02553.x.
- ↑ Sally A. Arif, PharmD, BCPS (Department of Pharmacy Practice, Chicago College of Pharmacy, Midwestern University, Downers Grove, Illinois, and Department of Pharmacy, Rush University Medical Center, Chicago, Illinois); and Henry Poon, PharmD, BCPS (Department of Pharmacy, James J. Peters VA Medical Center, Bronx, New York). Tadalafil: A Long-Acting Phosphodiesterase-5 Inhibitor for the Treatment of Pulmonary Arterial Hypertension. 2011;33:993–1004
- ↑ Galié N, Brundage BH, Ghofrani HA; et al. (2009). "Tadalafil therapy for pulmonary arterial hypertension". Circulation. 119: 2894–2903. doi:10.1161/circulationaha.108.839274.
- ↑ Lasker, George F; Maley, Jason H; Pankey, Edward A; Kadowitz, Philip J (2011-04-01). "Targeting soluble guanylate cyclase for the treatment of pulmonary hypertension". Expert review of respiratory medicine. 5 (2): 153–161. doi:10.1586/ers.11.9. ISSN 1747-6348. PMC 3108035. PMID 21510726.
- ↑ "2006 OPTN/SRTR Annual Report". US Scientific Registry of Transplant Recipients. 2006-05-01. Retrieved 2007-03-28.
- ↑ Cerveri, I; D’Armini, A M; Viganò, M (2003-04-01). "Pulmonary thromboendarterectomy almost 50 years after the first surgical attempts". Heart. 89 (4): 369–370. doi:10.1136/heart.89.4.369. ISSN 1355-6037. PMC 1769265. PMID 12639858.
- ↑ American College of Chest Physicians; American Thoracic Society (September 2013), "Five Things Physicians and Patients Should Question", Choosing Wisely: an initiative of the ABIM Foundation, American College of Chest Physicians and American Thoracic Society, retrieved 6 January 2013
- ↑ "Pulmonary Hypertension. About Pulmonary Hypertension | Patient". Patient. Retrieved 2015-12-30.
- ↑ McLaughlin VV, Sitbon O, Badesch DB, et al. (2005). "Survival with first-line bosentan in patients with primary pulmonary hypertension". Eur. Respir. J. 25 (2): 244–9. doi:10.1183/09031936.05.00054804. PMID 15684287.
- ↑ Benza RL; Miller DP; Barst RJ; Badesch DB; Frost AE; McGoon MD. (August 2012). "An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry.". Chest. 142 (2): 448–56. doi:10.1378/chest.11-1460. PMID 22281797. [FREE]
- ↑ Kaufman, Matthew H.; Latha Stead; Feig, Robert (2007). First aid for the obstetrics & gynecology clerkship. New York: McGraw-Hill, Medical Pub. Division. p. 100. ISBN 0-07-144874-8.
- ↑ Ghosh, Amit K. (2008). Mayo Clinic Internal Medicine Review: Eighth Edition (Mayo Clinic Internal Medicine Review). Informa Healthcare. p. 55. ISBN 1-4200-8478-X.
- ↑ British Journal of Anaesthesia: "Primary pulmonary hypertension in pregnancy; a role for novel vasodilators" March 19, 2011
- ↑ "Primary Pulmonary Hypertension: Practice Essentials, Background, Pathophysiology".
- ↑ York, Michael; Farber, Harrison W. (November 2011). "Pulmonary Hypertension: Screening and Evaluation in Scleroderma". Current Opinion in Rheumatology. 23 (6): 536–544. doi:10.1097/BOR.0b013e32834ba6a7. Retrieved 8 January 2016.
- ↑ Nannini, Carlotta. "Lung Disease in Rheumatoid Arthritis". MedScape.com. MedScape. Retrieved 31 December 2015.
- ↑ Mittoo, Shikha. "Pulmonary Manifestations of Systemic Lupus Erythematosus". MedScape.com. MedScape. Retrieved 31 December 2015.
- ↑ Lee, M (2007). "Pulmonary Hypertension in Sickle Cell Disease" (PDF). Clinical Advances in Hematology and Oncology. Retrieved 31 December 2015.
- ↑ Hoeper, Marius M.; Mayer, Eckhard; Simonneau, Gérald; Rubin, Lewis J. (2006-04-25). "Chronic Thromboembolic Pulmonary Hypertension". Circulation. 113 (16): 2011–2020. doi:10.1161/CIRCULATIONAHA.105.602565. ISSN 0009-7322. PMID 16636189.
- ↑ Minai, Omar A.; Chaouat, Ari; Adnot, Serge (2010-06-01). "Pulmonary hypertension in copd: Epidemiology, significance, and management: pulmonary vascular disease: the global perspective". Chest. 137 (6_suppl): 39S–51S. doi:10.1378/chest.10-0087. ISSN 0012-3692.
- ↑ Balachandran, Jay S.; Masa, Juan Fernando; Mokhlesi, Babak (2014-09-01). "Obesity Hypoventilation Syndrome Epidemiology and Diagnosis". Sleep medicine clinics. 9 (3): 341–347. doi:10.1016/j.jsmc.2014.05.007. ISSN 1556-407X. PMC 4210766. PMID 25360072.
- ↑ "Elaine Kaufman, famed Elaine's restaurateur, dies at age 81". Daily News. Retrieved 27 September 2016.
- ↑ "Why I Only Have One Kid, If You Insist on Asking". Yahoo News. Retrieved 27 September 2016.
- ↑ FOLKART, BURT A. (21 June 1990). "Ina Balin, 52; Movie and TV Actress Sought Lung Implant". Los Angeles Times. Retrieved 27 September 2016.
- ↑ Phull, Hardeep (July 26, 2016). "She's tethered to an oxygen tank, but her singing career is soaring". New York Post.
- ↑ Insdorf, Annette (November 10, 2013). "The Challenges of Chloe Temtchine". The Huffington Post.
- ↑ Bryant, Miranda. "Singer who has to take oxygen tank everywhere due to rare disorder". Daily Mail.
- ↑ Valadez, Brittany (8 January 2016). "Natalie Cole's cause of death revealed as heart failure from pulmonary hypertension". Daily Mail. Retrieved 21 November 2016.
Further reading
- Rubin LJ, Badesch DB (2005). "Evaluation and management of the patient with pulmonary arterial hypertension". Ann. Intern. Med. 143 (4): 282–92. doi:10.7326/0003-4819-143-4-200508160-00009. PMID 16103472.
- Abman, SH; Hansmann, G; Archer, SL; Ivy, DD; Adatia, I; Chung, WK; Hanna, BD; Rosenzweig, EB; Raj, JU; Cornfield, D; Stenmark, KR; Steinhorn, R; Thébaud, B; Fineman, JR; Kuehne, T; Feinstein, JA; Friedberg, MK; Earing, M; Barst, RJ; Keller, RL; Kinsella, JP; Mullen, M; Deterding, R; Kulik, T; Mallory, G; Humpl, T; Wessel, DL (3 November 2015). "Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society.". Circulation. 132: 2037–99. doi:10.1161/CIR.0000000000000329. PMID 26534956.
External links
| Wikimedia Commons has media related to Pulmonary hypertension. |
- Pulmonary Hypertension Association
- European Pulmonary Hypertension Association
- The Merck Manual Home Edition: Pulmonary Hypertension
- Pulmonary Arterial Hypertension database
- PH Central - the internet resource for Pulmonary Arterial Hypertension
- Webcast: The Changing World of Pulmonary Arterial Hypertension Therapies - American College of CHEST Physicians
- OMIM entries on Heritable Pulmonary Arterial Hypertension
- Pulmonary Hypertension Association of Australia