Pulmonary alveolar proteinosis
| Pulmonary alveolar proteinosis | |
|---|---|
 | |
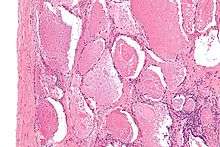
| Micrograph of pulmonary alveolar proteinosis, showing the characteristic airspace filling with focally dense globs referred to as chatter or dense bodies. H&E stain. | |
| Classification and external resources | |
| Specialty | pulmonology |
| ICD-10 | J84.0 |
| ICD-9-CM | 516.0 |
| OMIM | 265120 610913 610921 610910 |
| DiseasesDB | 29642 |
| MedlinePlus | 000114 |
| eMedicine | article/300615 |
| MeSH | D011649 |
Pulmonary alveolar proteinosis (abbreviated PAP), is a rare lung disease in which abnormal accumulation of pulmonary surfactant occurs within the alveoli, interfering with gas exchange. PAP can occur in a primary form or secondarily in the settings of malignancy (especially in myeloid leukemia), pulmonary infection, or environmental exposure to dusts or chemicals. Rare familial forms have also been recognized, suggesting a genetic component in some cases.
Signs and symptoms
The symptoms of PAP include:
The clinical course of PAP is unpredictable. Spontaneous remission is recognized; some patients have stable symptoms. Death may occur due to progression of PAP or due to the underlying disease associated with PAP. Individuals with PAP are more vulnerable to infection of the lung by bacteria or fungi.
Mechanism
Although the cause of PAP remains obscure, a major breakthrough in the understanding of the etiology of the disease came by the chance observation that mice bred for experimental study to lack a hematologic growth factor known as granulocyte-macrophage colony stimulating factor (GM-CSF) developed a pulmonary syndrome of abnormal surfactant accumulation resembling human PAP.[2]
The implications of this finding are still being explored, but significant progress was reported in February, 2007. Researchers in that report discussed the presence of anti-GM-CSF autoantibodies in patients with PAP, and duplicated that syndrome with the infusion of these autoantibodies into mice.[3]
Diagnosis
Chest x-rays of affected individuals typically reveal nonspecific alveolar opacities. Diagnosis is generally made by surgical or endoscopic biopsy of the lung, revealing the distinctive pathologic finding. The current gold standard of PAP diagnosis involves histopathological examination of alveolar specimens obtained from bronchoalveolar lavage and transbronchial lung biopsy.[4]
Microscopically, the distal air spaces are filled with a granular, eosinophilic material that is positive with the PAS stain and the PAS diastase stain. The main histomorphologic differential diagnosis is pulmonary edema, which does not have dense bodies.
An ELISA to measure antibodies against GM-CSF has been validated for routine clinical diagnosis of autoimmune PAP.[5]
Treatment
The first advance in the treatment of Pulmonary Alveolar Proteinosis came in November 1960,[6] when Dr. Jose Ramirez-Rivera at the Veterans' Administration Hospital in Baltimore applied repeated "segmental flooding" as a means of physically removing the accumulated alveolar material.[7]
The standard treatment for PAP is whole-lung lavage,[8][9] in which sterile fluid is instilled into the lung and then removed, along with the abnormal surfactant material. This is generally effective at ameliorating symptoms, often for prolonged periods. Since the mouse discovery noted above, the use of GM-CSF injections has also been attempted, with variable success. Lung transplantation can be performed in refractory cases.
Epidemiology
The disease is more common in males and in tobacco smokers.
In a recent epidemiologic study from Japan,[10] Autoimmune PAP has an incidence and prevalence higher than previously reported and is not strongly linked to smoking, occupational exposure, or other illnesses.
Endogenous lipoid pneumonia and non-specific interstitial pneumonitis has been seen prior to the development of PAP in a child.[11]
Clinical Research
PAP is one of the rare lung diseases currently being studied by The Rare Lung Diseases Consortium (RLDC). The RLDC is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), of the National Center for Advancing Translational Sciences (NCATS). The RLDC is dedicated to developing new diagnostics and therapeutics for patients with rare lung diseases, through collaboration between the NIH, patient organizations and clinical investigators.
Patient Registry
PAP patients, families, and caregivers are encouraged to join the NIH Rare Lung Diseases Consortium Contact Registry. This is a privacy protected site that provides up-to-date information for individuals interested in the latest scientific news, trials, and treatments related to rare lung diseases.
History
PAP was first described in 1958[12] by the physicians Samuel Rosen, Benjamin Castleman, and Averill Liebow.[13] In their case series published in the New England Journal of Medicine on June 7 of that year, they described 27 patients with pathologic evidence of periodic acid Schiff positive material filling the alveoli. This lipid rich material was subsequently recognized to be surfactant.
References
- ↑ Shah PL, Hansell D, Lawson PR, Reid KB, Morgan C (January 2000). "Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis". Thorax. 55 (1): 67–77. doi:10.1136/thorax.55.1.67. PMC 1745595
 . PMID 10607805.
. PMID 10607805. - ↑ Stanley E, Lieschke GJ, Grail D, et al. (June 1994). "Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology". Proc. Natl. Acad. Sci. U.S.A. 91 (12): 5592–6. doi:10.1073/pnas.91.12.5592. PMC 44042. PMID 8202532.
- ↑ Uchida K, Beck D, Yamamoto T, Berclaz P, Abe S, Staudt M, Carey B, Filippi M, Wert S, Denson L, Puchalski J, Hauck D, Trapnell B (2007). "GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis". N Engl J Med. 356 (6): 567–79. doi:10.1056/NEJMoa062505. PMID 17287477.
- ↑ Monisha Das, MD & Gary A. Salzman, MD (February 2010). "Pulmonary Alveolar Proteinosis: An Overview for Internists and Hospital Physicians". Hospital Practice. 38 (1): 43–49. doi:10.3810/hp.2010.02.277. PMID 20469623.
- ↑ Uchida K, Nakata K, Carey B, Chalk C, Suzuki T, Sakagami T, Koch DE, Stevens C, Inoue Y, Yamada Y, Trapnell BC (Jan 15, 2014). "Standardized serum GM-CSF autoantibody testing for the routine clinical diagnosis of autoimmune pulmonary alveolar proteinosis". J Immunol Methods. 402 (1-2): 57–70. doi:10.1016/j.jim.2013.11.011. PMID 24275678.
- ↑ Ramirez-Rivera J, Nyka W, McLaughlin J (1963). "Pulmonary Alveolar Proteinosis: Diagnostic Technics and Observations". New England Journal of Medicine. 268 (4): 165–71. doi:10.1056/NEJM196301242680401. PMID 13990655.
- ↑ Ramirez-Rivera J, Schultz RB, Dutton RE (1963). "Pulmonary Alveolar Proteinosis: A New Technique and Rational for Treatment". Archives of Internal Medicine. 112: 419–31. doi:10.1001/archinte.1963.03860030173021.
- ↑ Ceruti M, Rodi G, Stella GM, et al. (2007). "Successful whole lung lavage in pulmonary alveolar proteinosis secondary to lysinuric protein intolerance: a case report". Orphanet J Rare Dis. 2: 14. doi:10.1186/1750-1172-2-14. PMC 1845139. PMID 17386098.
- ↑ Menard KJ (April 2005). "Whole lung lavage in the treatment of pulmonary alveolar proteinosis". J. Perianesth. Nurs. 20 (2): 114–26. doi:10.1016/j.jopan.2005.01.005. PMID 15806528.
- ↑ Inoue Y, Trapnell BC, Tazawa R, et al. (April 2008). "Characteristics of a Large Cohort of Patients with Autoimmune Pulmonary Alveolar Proteinosis in Japan". Am. J. Respir. Crit. Care Med. 177 (7): 752–62. doi:10.1164/rccm.200708-1271OC. PMC 2720118. PMID 18202348.
- ↑ Antoon JW, Hernandez ML, Roehrs PA, Noah TL, Leigh MW, Byerley JS (2014). "Endogenous lipoid pneumonia preceding diagnosis of pulmonary alveolar proteinosis". Clin Respir J. doi:10.1111/crj.12197. PMID 25103284.
- ↑ Seymour JF, Presneill JJ (July 2002). "Pulmonary alveolar proteinosis: progress in the first 44 years". Am. J. Respir. Crit. Care Med. 166 (2): 215–35. doi:10.1164/rccm.2109105. PMID 12119235.
- ↑ Rosen SH, Castleman B, Liebow AA (1958). "Pulmonary alveolar proteinosis". N. Engl. J. Med. 258 (23): 1123–1142. doi:10.1056/NEJM195806052582301. PMID 13552931.
External links
- Pap Foundation/ The american foundation for patients affected by PAP
- EuPAPNet/ The european consortium for PAP
- ORPHANET/ The portal for rare diseases and orphan drugs
- Pulmonary Alveolar Proteinosis: An Overview for Internists and Hospital Physicians
