Neuromuscular junction

| Neuromuscular junctions | |
|---|---|
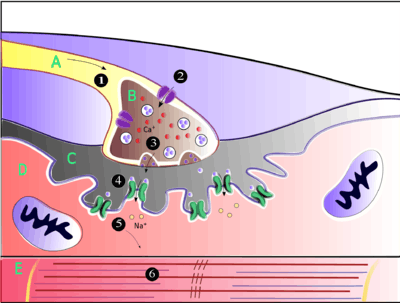
.jpg) Electron micrograph showing a cross section through the neuromuscular junction. T is the axon terminal, M is the muscle fiber. The arrow shows junctional folds with basal lamina. Active zones are visible on the tips between the folds. Scale is 0.3 µm. Source: NIMH | |
 Detailed view of a neuromuscular junction: | |
| Details | |
| Identifiers | |
| Latin | synapssis neuromuscularis; junctio neuromuscularis |
| TH | H2.00.06.1.02001 |
| FMA | 61803 |
A neuromuscular junction (or myoneural junction) is a chemical synapse formed by the contact between a motor neuron and a muscle fiber.[1] It is at the neuromuscular junction that a motor neuron is able to transmit a signal to the muscle fiber, causing muscle contraction.
Muscles require innervation to function—and even just to maintain muscle tone, avoiding atrophy. Synaptic transmission at the neuromuscular junction begins when an action potential reaches the presynaptic terminal of a motor neuron, which activates voltage-dependent calcium channels to allow calcium ions to enter the neuron. Calcium ions bind to sensor proteins (synaptotagmin) on synaptic vesicles, triggering vesicle fusion with the cell membrane and subsequent neurotransmitter release from the motor neuron into the synaptic cleft. In vertebrates, motor neurons release acetylcholine (ACh), a small molecule neurotransmitter, which diffuses across the synaptic cleft and binds to nicotinic acetylcholine receptors (nAChRs) on the cell membrane of the muscle fiber, also known as the sarcolemma. nAChRs are ionotropic receptors, meaning they serve as ligand-gated ion channels. The binding of ACh to the receptor can depolarize the muscle fiber, causing a cascade that eventually results in muscle contraction.
Neuromuscular junction diseases can be of genetic and autoimmune origin. Genetic disorders, such as Duchenne muscular dystrophy, can arise from mutated structural proteins that comprise the neuromuscular junction, whereas autoimmune diseases, such as myasthenia gravis, occur when antibodies are produced against nicotinic acetylcholine receptors on the sarcolemma.
Structure and function of the neuromuscular junction

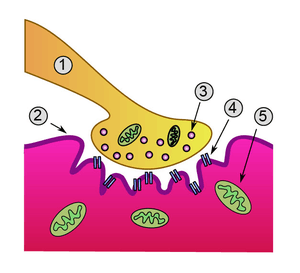
The neuromuscular junction differs from chemical synapses between neurons. Presynaptic motor axons stop 30 nanometers from the sarcolemma, the cell membrane of a muscle cell. This 30-nanometer space forms the synaptic cleft through which signalling molecules are released. The sarcolemma has invaginations called postjunctional folds, which increase the surface area of the membrane exposed to the synaptic cleft.[2] These postjunctional folds form what is referred to as the motor endplate, which possess nicotinic acetylcholine receptors (nAChRs) at a density of 10,000 receptors/micrometer2 in skeletal muscle.[3] The presynaptic axons form bulges called terminal boutons (or presynaptic terminals) that project into the postjunctional folds of the sarcolemma. The presynaptic terminals have active zones that contain vesicles, also called quanta, full of acetylcholine molecules. These vesicles can fuse with the presynaptic membrane and release ACh molecules into the synaptic cleft via exocytosis after depolarization.[2] AChRs are localized opposite the presynaptic terminals by protein scaffolds at the postjunctional folds of the sarcolemma. Dystrophin, a structural protein, connects the sarcomere, sarcolemma, and extracellular matrix components. Rapsyn is another protein that docks AChRs and structural proteins to the cytoskeleton. Also present is the receptor tyrosine kinase protein MuSK, a signaling protein involved in the development of the neuromuscular junction, which is also held in place by rapsyn.[2]
Mechanism of action
The neuromuscular junction is where a neuron activates a muscle to contract. Upon the arrival of an action potential at the presynaptic neuron terminal, voltage-dependent calcium channels open and Ca2+ ions flow from the extracellular fluid into the presynaptic neuron's cytosol. This influx of Ca2+ causes neurotransmitter-containing vesicles to dock and fuse to the presynaptic neuron's cell membrane through SNARE proteins. Fusion of the vesicular membrane with the presynaptic cell membrane results in the emptying of the vesicle's contents (acetylcholine) into the synaptic cleft, a process known as exocytosis. Acetylcholine diffuses into the synaptic cleft and can bind to the nicotinic acetylcholine receptors on the motor endplate.
Acetylcholine receptors at the neuromuscular junction
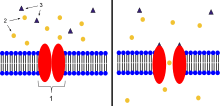
2. Ions
3. Ligand (such as acetylcholine)
When ligands bind to the receptor, the ion channel portion of the receptor opens, allowing ions to pass across the cell membrane.
Acetylcholine is a neurotransmitter synthesized from dietary choline and acetyl-CoA (ACoA), and is involved in the stimulation of muscle tissue in vertebrates as well as in some invertebrate animals. In vertebrate animals, the acetylcholine receptor subtype that is found at the neuromuscular junction of skeletal muscles is the nicotinic acetylcholine receptor (nAChR), which is a ligand-gated ion channel. Each subunit of this receptor has a characteristic “cys-loop,” which is composed of a cysteine residue followed by 13 amino acid residues and another cysteine residue. The two cysteine residues form a disulfide linkage which results in the “cys-loop” receptor that is capable of binding acetylcholine and other ligands. These cys-loop receptors are found only in eukaryotes, but prokaryotes possess ACh receptors with similar properties.[3] Not all species use a cholinergic neuromuscular junction; e.g. crayfish and fruit flies have a glutamatergic neuromuscular junction.[2]
AChRs at the skeletal neuromuscular junction form heteropentamers composed of two α, one β, one ɛ, and one δ subunits.[4] When a single ACh ligand binds to one of the α subunits of the ACh receptor it induces a conformational change at the interface with the second AChR α subunit. This conformational change results in the increased affinity of the second α subunit for a second ACh ligand. AChRs therefore exhibit a sigmoidal dissociation curve due to this cooperative binding .[3] The presence of the inactive, intermediate receptor structure with a single-bound ligand keeps ACh in the synapse that might otherwise be lost by cholinesterase hydrolysis or diffusion. The persistence of these ACh ligands in the synapse can cause a prolonged post-synaptic response.[5]
Development of the neuromuscular junction
The development of the neuromuscular junction requires signaling from both the motor neuron's terminal and the muscle cell's central region, During development, muscle cells produce acetylcholine receptors (AChRs) and express them in the central regions in a process called prepatterning. Agrin, a heparin proteoglycan, and MuSK kinase are thought to help stabilize the accumulation of AChR in the central regions of the myocyte. MuSK is a receptor tyrosine kinase — meaning that it induces cellular signaling by binding phosphate molecules to self regions like tyrosines, and to other targets in the cytoplasm.[6] Upon activation by its ligand agrin, MuSK signals via two proteins called "Dok-7" and "rapsyn", to induce "clustering" of acetylcholine receptors.[7] ACh release by developing motor neurons produces postsynaptic potentials in the muscle cell that positively reinforces the localization and stabilization of the developing neuromuscular junction.[8]
These findings were demonstrated in part by mouse "knockout" studies. In mice which are deficient for either agrin or MuSK, the neuromuscular junction does not form. Further, mice deficient in Dok-7 did not form either acetylcholine receptor clusters or neuromuscular synapses.[9]
The development of neuromuscular junctions is mostly studied in model organisms, such as rodents. In addition, in 2015 an all-human neuromuscular junction has been created in vitro using human embryonic stem cells and somatic muscle stem cells.[10] In this model presynaptic motor neurons are activated by optogenetics and in response synaptically connected muscle fibers twitch upon light stimulation.
Research methods
Joel del Castillo and Bernard Katz used ionophoresis to determine the location and density of nicotinic acetylcholine receptors (nAChRs) at the neuromuscular junction. With this technique, a microelectrode was placed inside the motor endplate of the muscle fiber, and a micropipette filled with acetylcholine (ACh) is placed directly in front of the endplate in the synaptic cleft. A positive voltage was applied to the tip of the micropipette, which caused a burst of positively charged ACh molecules to be released from the pipette. These ligands flowed into the space representing the synaptic cleft and bound to AChRs. The intracellular microelectrode monitored the amplitude of the depolarization of the motor endplate in response to ACh binding to nicotinic (ionotropic) receptors. Katz and del Castillo showed that the amplitude of the depolarization (excitatory postsynaptic potential) depended on the proximity of the micropipette releasing the ACh ions to the endplate. The farther the micropipette was from the motor endplate, the smaller the depolarization was in the muscle fiber. This allowed the researchers to determine that the nicotinic receptors were localized to the motor endplate in high density.[2][3]
Toxins are also used to determine the location of acetylcholine receptors at the neuromuscular junction. α-Bungarotoxin is a toxin found in the snake species Bungarus multicinctus that acts as an ACh antagonist and binds to AChRs irreversibly. By coupling assayable enzymes such as horseradish peroxidase (HRP) or fluorescent proteins such as green fluorescent protein (GFP) to the α-bungarotoxin, AChRs can be visualized and quantified.[2]
Toxins that affect the neuromuscular junction
Nerve gases
Nerve gases and liquor damage this area too.
Botulinum toxin
Botulinum toxin (aka botulinum neurotoxin, BoNT, and sold under the trade name Botox) inhibits the release of acetylcholine at the neuromuscular junction by interfering with SNARE proteins.[2] This toxin crosses into the nerve terminal through the process of endocytosis and subsequently interferes with SNARE proteins, which are necessary for ACh release. By doing so, it induces a transient paralysis and chemical denervation localized to the striated muscle that it has affected. The inhibition of the ACh release does not set in until approximately two weeks after the injection is made. Three months after the inhibition occurs, neuronal activity begins to regain partial function, and six months, complete neuronal function is regained.[11]
Tetanus toxin
Tetanus toxin, also known as Tetanospasmin is a potent neurotoxin produced by Clostridium Tetani and causes the disease state, tetanus. The LD50 of this toxin has been measured to be approximately 1 ng/kg, making it second only to Botulinum toxin D as the deadliest toxin in the world. It functions very similarly to botunlinum neurotoxin (BoNT) by attaching and endocytosing into the presynaptic nerve terminal and interfering with SNARE protein complexes. It differs from BoNT in a few ways, most apparently in its end state, wherein tetanospasmin demonstrates a rigid / spastic paralysis as opposed to the flaccid paralysis demonstrated with BoNT.
Latrotoxin
Latrotoxin (α-Latrotoxin) found in venom of widow spiders also affects the neuromuscular junction by causing the release of acetylcholine from the presynaptic cell. Mechanisms of action include binding to receptors on the presynaptic cell activating the IP3/DAG pathway and release of calcium from intracellular stores and pore formation resulting in influx of calcium ions directly. Either mechanism causes increased calcium in presynaptic cell, which then leads to release of synaptic vesicles of acetylcholine. Latrotoxin causes pain, muscle contraction and if untreated potentially paralysis and death.
Snake venom
Snake venoms act as toxins at the neuromuscular junction and can induce weakness and paralysis. Venoms can act as both presynaptic and postsynaptic neurotoxins.[12]
Presynaptic neurotoxins, commonly known as β-neurotoxins, affect the presynaptic regions of the neuromuscular junction. The majority of these neurotoxins act by inhibiting the release of neurotransmitters, such as acetylcholine, into the synapse between neurons. However, some of these toxins have also been known to enhance neurotransmitter release. Those that inhibit neurotransmitter release create a neuromuscular blockade that prevents signaling molecules from reaching their postsynaptic target receptors. In doing so, the victim of these snake bite suffer from profound weakness. Such neurotoxins do not respond well to anti-venoms. After one hour of inoculation of these toxins, including notexin and taipoxin, many of the affected nerve terminals show signs of irreversible physical damage, leaving them devoid of any synaptic vesicles.[12]
Postsynaptic neurotoxins, otherwise known as α-neurotoxins, act oppositely to the presynaptic neurotoxins by binding to the postsynaptic acetylcholine receptors. This prevents interaction between the acetylcholine released by the presynaptic terminal and the receptors on the postsynaptic cell. In effect, the opening of sodium channels associated with these acetylcholine receptors is prohibited, resulting in a neuromuscular blockade, similar to the effects seen due to presynaptic neurotoxins. This causes paralysis in the muscles involved in the affected junctions. Unlike presynaptic neurotoxins, postsynaptic toxins are more easily affected by anti-venoms, which accelerate the dissociation of the toxin from the receptors, ultimately causing a reversal of paralysis. These neurotoxins experimentally and qualitatively aid in the study of acetylcholine receptor density and turnover, as well as in studies observing the direction of antibodies toward the affected acetylcholine receptors in patients diagnosed with myasthenia gravis.[12]
Diseases
Any disorder that compromises the synaptic transmission between a motor neuron and a muscle cell is categorized under the umbrella term of neuromuscular diseases. These disorders can be inherited or acquired and can vary in their severity and mortality. In general, most of these disorders tend to be caused by mutations or autoimmune disorders. Autoimmune disorders, in the case of neuromuscular diseases, tend to be humoral mediated, B cell mediated, and result in an antibody improperly created against a motor neuron or muscle fiber protein that interferes with synaptic transmission or signaling.
Autoimmune
Myasthenia gravis
Myasthenia gravis is an autoimmune disorder where the body makes antibodies against either the acetylcholine receptor (AchR) (in 80% of cases), or against postsynaptic muscle-specific kinase (MuSK) (0–10% of cases). In seronegative myasthenia gravis low density lipoprotein receptor-related protein 4 is targeted by IgG1, which acts as a competitive inhibitor of its ligand, preventing the ligand from binding its receptor. It is not known if seronegative myasthenia gravis will respond to standard therapies.[13]
Neonatal MG
Neonatal MG is an autoimmune disorder that affects 1 in 8 children born to mothers who have been diagnosed with Myasthenia gravis (MG). MG can be transferred from the mother to the fetus by the movement of AChR antibodies through the placenta. Signs of this disease at birth include weakness, which responds to anticholinesterase medications, as well as fetal akinesia, or the lack of fetal movement. This form of the disease is transient, lasting for about three months. However, in some cases, neonatal MG can lead to other health effects, such as arthrogryposis and even fetal death. These conditions are thought to be initiated when maternal AChR antibodies are directed to the fetal AChR and can last until the 33rd week of gestation, when the γ subunit of AChR is replaced by the ε subunit.[14]
Lambert-Eaton myasthenic syndrome
Lambert-Eaton myasthenic syndrome (LEMS) is an autoimmune disorder that affects the presynaptic portion of the neuromuscular junction. This rare disease can be marked by a unique triad of symptoms: proximal muscle weakness, autonomic dysfunction, and areflexia.[15] Proximal muscle weakness is a product of pathogenic autoantibodies directed against P/Q-type voltage-gated calcium channels, which in turn leads to a reduction of acetylcholine release from motor nerve terminals on the presynaptic cell. Examples of autonomic dysfunction caused by LEMS include erectile dysfunction in men, constipation, and, most commonly, dry mouth. Less common dysfunctions include dry eyes and altered perspiration. Areflexia is a condition in which tendon reflexes are reduced and it may subside temporarily after a period of exercise.[16]
50–60% of the patients that are diagnosed with LEMS also have present an associated tumor, which is typically small-cell lung carcinoma (SCLC). This type of tumor also expresses voltage-gated calcium channels.[16] Oftentimes, LEMS also occurs alongside myasthenia gravis.[15]
Treatment for LEMS consists of using 3,4-diaminopyridine as a first measure, which serves to increase the compound muscle action potential as well as muscle strength by lengthening the time that voltage-gated calcium channels remain open after blocking voltage-gated potassium channels. In the US, treatment with 3,4-diaminopyridine for eligible LEMS patients is available at no cost under an expanded access program.[17][18] Further treatment includes the use of prednisone and azathioprine in the event that 3,4-diaminopyridine does not aid in treatment.[16]
Neuromyotonia
Neuromyotonia (NMT), otherwise known as Isaac’s syndrome, is unlike many other diseases present at the neuromuscular junction. Rather than causing muscle weakness, NMT leads to the hyperexcitation of motor nerves. NMT causes this hyperexcitation by producing longer depolarizations by down-regulating voltage-gated potassium channels, which causes greater neurotransmitter release and repetitive firing. This increase in rate of firing leads to more active transmission and as a result, greater muscular activity in the affected individual. NMT is also believed to be of autoimmune origin due to its associations with autoimmune symptoms in the individual affected.[14]
Genetic
Congenital myasthenic syndromes
Congenital myasthenic syndromes (CMS) are very similar to both MG and LEMS in their functions, but the primary difference between CMS and those diseases is that CMS is of genetic origins. Specifically, these syndromes are diseases incurred due to mutations, typically recessive, in 1 of at least 10 genes that affect presynaptic, synaptic, and postsynaptic proteins in the neuromuscular junction. Such mutations usually arise in the ε-subunit of AChR,[14] thereby affecting the kinetics and expression of the receptor itself. Single nucleotide substitutions or deletions may cause loss of function in the subunit. Other mutations, such as those affecting acetylcholinesterase and acetyltransferase, can also cause the expression of CMS, with the latter being associated specifically with episodic apnea.[19] These syndromes can present themselves at different times within the life of an individual. They may arise during the fetal phase, causing fetal akinesia, or the perinatal period, during which certain conditions, such as arthrogryposis, ptosis, hypotonia, ophthalmoplegia, and feeding or breathing difficulties, may be observed. They could also activate during adolescence or adult years, causing the individual to develop slow-channel syndrome.[14]
Treatment for particular subtypes of CMS (postsynaptic fast-channel CMS)[20][21] is similar to treatment for other neuromuscular disorders. 3,4-Diaminopyridine, the first-line treatment for LEMS, is under development as an orphan drug for CMS[22] in the US, and available to eligible patients under an expanded access program at no cost.[17][18]
Bulbospinal muscular atrophy
Bulbospinal muscular atrophy, also known as Kennedy’s disease, is a rare recessive trinucleotide, polyglutamine disorder that is linked to the X chromosome. Because of its linkage to the X chromosome, it is typically transmitted through females. However, Kennedy’s disease is only present in adult males and the onset of the disease is typically later in life. This disease is specifically caused by the expansion of a CAG-tandem repeat in exon 1 found on the androgen-receptor (AR) gene on chromosome Xq11-12. Poly-Q-expanded AR accumulates in the nuclei of cells, where it begins to fragment. After fragmentation, degradation of the cell begins, leading to a loss of both motor neurons and dorsal root ganglia.[23]
Symptoms of Kennedy’s disease include weakness and wasting of the facial bulbar and extremity muscles, as well as sensory and endocrinological disturbances, such as gynecomastia and reduced fertility. Other symptoms include elevated testosterone and other sexual hormone levels, development of hyper-CK-emia, abnormal conduction through motor and sensory nerves, and neuropathic or in rare cases myopathic alterations on biopsies of muscle cells.[23]
Duchenne muscular dystrophy
Duchenne muscular dystrophy is an X-linked genetic disorder that results in the absence of the structural protein dystrophin at the neuromuscular junction. It affects 1 in 3,600–6,000 males and frequently causes death by the age of 30. The absence of dystrophin causes muscle degeneration, and patients present with the following symptoms: abnormal gait, hypertrophy in the calf muscles, and elevated creatine kinase. If left untreated, patients may suffer from respiratory distress, which can lead to death.[24]
See also
External links
- Histology image: 21501lca – Histology Learning System at Boston University
Further reading
- Kandel, ER; Schwartz JH; Jessell TM. (2000). Principles of Neural Science (4th ed.). New York: McGraw-Hill. ISBN 0-8385-7701-6.
- Nicholls, J.G.; A.R. Martin; B.G. Wallace; P.A. Fuchs (2001). From Neuron to Brain (4th ed.). Sunderland, MA.: Sinauer Associates. ISBN 0-87893-439-1.
- Engel, A.G. (2004). Myology (3rd ed.). New York: McGraw Hill Professional. ISBN 0-07-137180-X.
References
- ↑ Levitan, Irwin; Kaczmarek, Leonard (August 19, 2015). "Intercellular communication". The Neuron: Cell and Molecular Biology (4th ed.). New York, NY: Oxford Univerty Press. pp. 153–328. ISBN 0199773890.
- 1 2 3 4 5 6 7 Nicholls, John G.,; A. Robert Martin; Paul A. Fuchs; David A. Brown; Matthew E. Diamond; David A. Weisblat (2012). From Neuron to Brain (5th ed.). Sunderland: Sinauer Associates.
- 1 2 3 4 Sine SM (July 2012). "End-plate acetylcholine receptor: structure, mechanism, pharmacology, and disease". Physiol. Rev. 92 (3): 1189–234. doi:10.1152/physrev.00015.2011. PMC 3489064
 . PMID 22811427.
. PMID 22811427. - ↑ miller's anaesthesia, 7th edition.
- ↑ Scuka M, Mozrzymas JW (1992). "Postsynaptic potentiation and desensitization at the vertebrate end-plate receptors". Prog. Neurobiol. 38 (1): 19–33. doi:10.1016/0301-0082(92)90033-B. PMID 1736323.
- ↑ Valenzuela D, Stitt T, DiStefano P, Rojas E, Mattsson K, Compton D, Nuñez L, Park J, Stark J, Gies D (1995). "Receptor tyrosine sinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury". Neuron. 15 (3): 573–84. doi:10.1016/0896-6273(95)90146-9. PMID 7546737.
- ↑ Glass D, Bowen D, Stitt T, Radziejewski C, Bruno J, Ryan T, Gies D, Shah S, Mattsson K, Burden S, DiStefano P, Valenzuela D, DeChiara T, Yancopoulos G (1996). "Agrin acts via a MuSK receptor complex". Cell. 85 (4): 513–23. doi:10.1016/S0092-8674(00)81252-0. PMID 8653787.
- ↑ Witzemann V (November 2006). "Development of the neuromuscular junction". Cell Tissue Res. 326 (2): 263–71. doi:10.1007/s00441-006-0237-x. PMID 16819627.
- ↑ Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, Kubo S, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y (2006). "The muscle protein Dok-7 is essential for neuromuscular synaptogenesis". Science. 312 (5781): 1802–5. doi:10.1126/science.1127142. PMID 16794080.
- ↑ Steinbeck, JA; Jaiswal, MK; Calder, EL; Kishinevsky, S; Weishaupt, A; Toyka, KV; Goldstein, PA; Studer, L (7 January 2016). "Functional Connectivity under Optogenetic Control Allows Modeling of Human Neuromuscular Disease.". Cell stem cell. 18 (1): 134–43. doi:10.1016/j.stem.2015.10.002. PMID 26549107.
- ↑ Papapetropoulos S, Singer C (April 2007). "Botulinum toxin in movement disorders". Semin Neurol. 27 (2): 183–94. doi:10.1055/s-2007-971171. PMID 17390263.
- 1 2 3 Lewis RL, Gutmann L (June 2004). "Snake venoms and the neuromuscular junction". Semin Neurol. 24 (2): 175–9. doi:10.1055/s-2004-830904. PMID 15257514.
- ↑ Finsterer J, Papić L, Auer-Grumbach M (October 2011). "Motor neuron, nerve, and neuromuscular junction disease". Curr. Opin. Neurol. 24 (5): 469–74. doi:10.1097/WCO.0b013e32834a9448. PMID 21825986.
- 1 2 3 4 Newsom-Davis J (July 2007). "The emerging diversity of neuromuscular junction disorders". Acta Myol. 26 (1): 5–10. PMC 2949330. PMID 17915563.
- 1 2 Luigetti M, Modoni A, Lo Monaco M (October 2012). "Low rate repetitive nerve stimulation in Lambert-Eaton myasthenic syndrome: Peculiar characteristics of decremental pattern from a single-centre experience". Clin Neurophysiol. 124 (4): 825–6. doi:10.1016/j.clinph.2012.08.026. PMID 23036181.
- 1 2 3 Titulaer MJ, Lang B, Verschuuren JJ (December 2011). "Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies". Lancet Neurol. 10 (12): 1098–107. doi:10.1016/S1474-4422(11)70245-9. PMID 22094130.
- 1 2 , Muscular Dystrophy Association Press Release
- 1 2 , Rare Disease Report
- ↑ Harper CM (March 2004). "Congenital myasthenic syndromes". Semin Neurol. 24 (1): 111–23. doi:10.1055/s-2004-829592. PMID 15229798.
- ↑ Engel AG, et al. (April 2015). "Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment.". Lancet Neurol. 14 (4): 420–34. doi:10.1016/S1474-4422(14)70201-7. PMID 25792100.
- ↑ Engel AG, et al. "New horizons for congenital myasthenic syndromes.". Ann N Y Acad Sci. 1275: 1275:54–62. doi:10.1111/j.1749-6632.2012.06803.x. PMC 3546605. PMID 23278578.
- ↑ , FDA orphan drug designation
- 1 2 Finsterer J (November 2010). "Perspectives of Kennedy's disease". J. Neurol. Sci. 298 (1–2): 1–10. doi:10.1016/j.jns.2010.08.025. PMID 20846673.
- ↑ Beytía Mde L, Vry J, Kirschner J (May 2012). "Drug treatment of Duchenne muscular dystrophy: available evidence and perspectives". Acta Myol. 31 (1): 4–8. PMC 3440798. PMID 22655510.