Ligand (biochemistry)
In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. In protein-ligand binding, the ligand is usually a molecule which produces a signal by binding to a site on a target protein. The binding typically results in a change of conformation of the target protein. In DNA-ligand binding studies, the ligand can be a small molecule, ion,[1] or protein[2] which binds to the DNA double helix. The relationship between ligand and binding partner is a function of charge, hydrophobicity, and molecular structure. The instance of binding occurs over an infinitesimal range of time and space, so the rate constant is usually a very small number.
Binding occurs by intermolecular forces, such as ionic bonds, hydrogen bonds and Van der Waals forces. The association of docking is actually reversible through dissociation. Measurably irreversible covalent bonding between a ligand and target molecule is atypical in biological systems. In contrast to the definition of ligand in metalorganic and inorganic chemistry, in biochemistry it is ambiguous whether the ligand generally binds at a metal site, as is the case in hemoglobin. In general, the interpretation of ligand is contextual with regards to what sort of binding has been observed. The etymology stems from ligare, which means 'to bind'.
Ligand binding to a receptor protein alters the chemical conformation by affecting the three-dimensional shape orientation. The conformation of a receptor protein composes the functional state. Ligands include substrates, inhibitors, activators, and neurotransmitters. The rate of binding is called affinity, and this measurement typifies a tendency or strength of the effect. Binding affinity is actualized not only by host-guest interactions, but also by solvent effects that can play a dominant, steric role which drives non-covalent binding in solution.[3] The solvent provides a chemical environment for the ligand and receptor to adapt, and thus accept or reject each other as partners.
Radioligands are radioisotope labeled compounds are used in vivo as tracers in PET studies and for in vitro binding studies.
Receptor/ligand binding affinity
The interaction of most ligands with their binding sites can be characterized in terms of a binding affinity. In general, high-affinity ligand binding results from greater intermolecular force between the ligand and its receptor while low-affinity ligand binding involves less intermolecular force between the ligand and its receptor. In general, high-affinity binding results in a higher degree of occupancy for the ligand at its receptor binding site than is the case for low-affinity binding; the residence time (lifetime of the receptor-ligand complex) does not correlate. High-affinity binding of ligands to receptors is often physiologically important when some of the binding energy can be used to cause a conformational change in the receptor, resulting in altered behavior of an associated ion channel or enzyme.
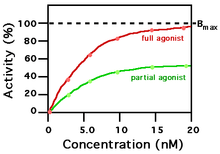
A ligand that can bind to a receptor, alter the function of the receptor, and trigger a physiological response is called an agonist for that receptor. Agonist binding to a receptor can be characterized both in terms of how much physiological response can be triggered and in terms of the concentration of the agonist that is required to produce the physiological response. High-affinity ligand binding implies that a relatively low concentration of a ligand is adequate to maximally occupy a ligand-binding site and trigger a physiological response. The lower the Ki concentration is, the more likely there will be a chemical reaction between the pending ion and the receptive antigen. Low-affinity binding (high Ki level) implies that a relatively high concentration of a ligand is required before the binding site is maximally occupied and the maximum physiological response to the ligand is achieved. In the example shown to the right, two different ligands bind to the same receptor binding site. Only one of the agonists shown can maximally stimulate the receptor and, thus, can be defined as a full agonist. An agonist that can only partially activate the physiological response is called a partial agonist. In this example, the concentration at which the full agonist (red curve) can half-maximally activate the receptor is about 5 x 10−9 Molar (nM = nanomolar). Ligands that bind to a receptor but fail to activate the physiological response are receptor antagonists.
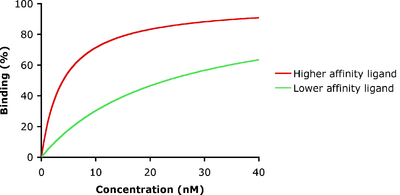
In the example shown to the left, ligand-binding curves are shown for two ligands with different binding affinities. Ligand binding is often characterized in terms of the concentration of ligand at which half of the receptor binding sites are occupied, known as the IC50, which is related to but different from the dissociation constant. The ligand illustrated by the red curve has a higher binding affinity and smaller Kd than the ligand illustrated by the green curve. If these two ligands were present at the same time, more of the higher-affinity ligand would be bound to the available receptor binding sites. This is how carbon monoxide can compete with oxygen in binding to hemoglobin, resulting in carbon monoxide poisoning.
Binding affinity is most commonly determined using a radiolabeled ligand, known as a tagged ligand. Homologous competitive binding experiments involve binding competition between a tagged ligand and an untagged ligand.[4] Non-labelled methods such as surface plasmon resonance, dual polarization interferometry and Multi-Parametric Surface Plasmon Resonance (MP-SPR) can not only quantify the affinity from concentration based assays; but also from the kinetics of association and dissociation, and in the later cases, the conformational change induced upon binding. MP-SPR also enables measurements in high saline dissociation buffers thanks to a unique optical setup. Microscale Thermophoresis (MST), an immobilization-free method[5] was developed. This method allows the determination of the binding affinity without any limitation to the ligand's molecular weight.[6]
For the use of statistical mechanics in a quantitative study of the ligand-receptor binding affinity, see the comprehensive article[7] on the configurational partition function.
Drug potency and binding affinity
Binding affinity data alone does not determine the overall potency of a drug. Potency is a result of the complex interplay of both the binding affinity and the ligand efficacy. Ligand efficacy refers to the ability of the ligand to produce a biological response upon binding to the target receptor and the quantitative magnitude of this response. This response may be as an agonist, antagonist, or inverse agonist, depending on the physiological response produced.[8]
Selective and non-selective
Selective ligands have a tendency to bind to very limited kinds of receptor, whereas non-selective ligands bind to several types of receptors. This plays an important role in pharmacology, where drugs that are non-selective tend to have more adverse effects, because they bind to several other receptors in addition to the one generating the desired effect.
Bivalent ligand
Bivalent ligands consist of two connected molecules as ligands, and are used in scientific research to detect receptor dimers and to investigate their properties. Bivalent ligands are usually large and tend not to be ‘drug-like’, limiting their applicability in clinical settings.[9][10] See Lipinski’s rule of five.
Privileged scaffold
A privileged scaffold[11] is a molecular framework or chemical moiety that is statistically recurrent among known drugs or among a specific array of biologically active compounds. These privileged elements[12] can be used as a basis for designing new active biological compounds or compound libraries.
Methods used to study binding
Main methods to study protein–ligand interactions are principal hydrodynamic and calorimetric techniques, and principal spectroscopic and structural methods such as
- Fourier transform spectroscopy
- Raman spectroscopy
- Fluorescence spectroscopy
- Circular dichroism
- Nuclear magnetic resonance
- Mass spectrometry
- Atomic force microscope
- Paramagnetic probes
- Dual polarisation interferometry
- Multi-parametric surface plasmon resonance
Other techniques include: fluorescence intensity, bimolecular fluorescence complementation, FRET (fluorescent resonance energy transfer) / FRET quenching surface plasmon resonance, bio-layer interferometry, Coimmunopreciptation indirect ELIS, equilibrium dialysis, gel electrophoresis, far western blot, fluorescence polarization anisotropy, electron paramagnetic resonance, microscale thermophoresis
The dramatically increased computing power of supercomputers and personal computers has made it possible to study protein–ligand interactions also by means of computational chemistry. For example, a worldwide grid of well over a million ordinary PCs was harnessed for cancer research in the project grid.org, which ended in April 2007. Grid.org has been succeeded by similar projects such as World Community Grid, Human Proteome Folding Project, Compute Against Cancer and Folding@Home.
See also
- Agonist
- Schild regression
- Allosteric regulation
- Ki Database
- Docking@Home
- GPUGRID.net
- DNA binding ligand
Notes
References
- ↑ Teif V.B. (2005). "Ligand-induced DNA condensation: choosing the model". Biophysical Journal. 89 (4): 2574–2587. doi:10.1529/biophysj.105.063909. PMC 1366757
 . PMID 16085765.
. PMID 16085765. - ↑ Teif VB, Rippe K (2010). "Statistical-mechanical lattice models for protein-DNA binding in chromatin.". Journal of Physics: Condensed Matter. 22 (41): 414105. doi:10.1088/0953-8984/22/41/414105. PMID 21386588.
- ↑ Baron, Riccardo; Setny, Piotr; Andrew Mccammon, J. (2010). "Water in Cavity-Ligand Recognition". Journal of the American Chemical Society. 132 (34): 12091–12097. doi:10.1021/ja1050082. PMC 2933114. PMID 20695475.
- ↑ See Homologous competitive binding curves, A complete guide to nonlinear regression, curvefit.com.
- ↑ Baaske P, Wienken CJ, Reineck P, Duhr S, Braun D (Feb 2010). "Optical Thermophoresis quantifies Buffer dependence of Aptamer Binding". Angew. Chem. Int. Ed. 49 (12): 1–5. doi:10.1002/anie.200903998. PMID 20186894. Lay summary – Phsyorg.com.
- ↑ Wienken CJ, et al. (2010). "Protein-binding assays in biological liquids using microscale thermophoresis". Nature Communications. 1 (7): 100. Bibcode:2010NatCo...1E.100W. doi:10.1038/ncomms1093. PMID 20981028.
- ↑ Vu-Quoc, L., Configuration integral (statistical mechanics), 2008. this wiki site is down; see this article in the web archive on 2012 April 28.
- ↑ Kenakin, Terrance P. (November 2006). A pharmacology primer: theory, applications, and methods. Academic Press. p. 79. ISBN 978-0-12-370599-0.
- ↑ Shonberg, Jeremy; Scammells, Peter J.; Capuano, Ben (June 2011). "Design strategies for bivalent ligands targeting GPCRs". ChemMedChem. 6 (6): 963–74. doi:10.1002/cmdc.201100101. PMID 21520422.
- ↑ Berque-Bestel, I; Lezoualc'h, F; Jockers, R (December 2008). "Bivalent ligands as specific pharmacological tools for G protein-coupled receptor dimers". Curr Drug Discov Technol. 5 (4): 312–8. doi:10.2174/157016308786733591. PMID 19075611.
- ↑ Welsch, ME; Snyder, SA; Stockwell, BR (2010). "Privileged scaffolds for library design and drug discovery". Curr Opin Chem Biol. 14: 347–61. doi:10.1016/j.cbpa.2010.02.018. PMC 2908274. PMID 20303320.
- ↑ Kombarov, R; Altieri, A; Genis, D; Kirpichenok, M; Kochubey, V; Rakitina, N; Titarenko, Z (2010). "Bio Cores: Identification of a drug/natural product-based privileged structural motif for small-molecule lead discovery". Molecular Diversity. 14 (1): 193–200. doi:10.1007/s11030-009-9157-5. PMID 19468851.
External links
- BindingDB, a public database of measured protein-ligand binding affinities.
- BioLiP, a comprehensive database for ligand-protein interactions.