Apoptosis
| Apoptosis | |
|---|---|
|
An etoposide-treated DU145 prostate cancer cell exploding into a cascade of apoptotic bodies. The sub images were extracted from a 61-hour time-lapse microscopy video, created using quantitative phase-contrast microscopy. The optical thickness is color-coded. With increasing thickness, color changes from gray to yellow, red, purple and finally black.[1] See the video at The Cell: An Image Library | |

Apoptosis (from Ancient Greek ἀπόπτωσις "falling off") is a process of programmed cell death that occurs in multicellular organisms.[2] Biochemical events lead to characteristic cell changes (morphology) and death. These changes include blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, chromosomal DNA fragmentation, and global mRNA decay. Between 50 and 70 billion cells die each day due to apoptosis in the average human adult.[lower-alpha 1] For an average child between the ages of 8 and 14, approximately 20 billion to 30 billion cells die a day.[4]
In contrast to necrosis, which is a form of traumatic cell death that results from acute cellular injury, apoptosis is a highly regulated and controlled process that confers advantages during an organism's lifecycle. For example, the separation of fingers and toes in a developing human embryo occurs because cells between the digits undergo apoptosis. Unlike necrosis, apoptosis produces cell fragments called apoptotic bodies that phagocytic cells are able to engulf and quickly remove before the contents of the cell can spill out onto surrounding cells and cause damage.[5]
Because apoptosis cannot stop once it has begun, it is a highly regulated process. Apoptosis can be initiated through one of two pathways. In the intrinsic pathway the cell kills itself because it senses cell stress, while in the extrinsic pathway the cell kills itself because of signals from other cells. Both pathways induce cell death by activating caspases, which are proteases, or enzymes that degrade proteins. The two pathways both activate initiator caspases, which then activate executioner caspases, which then kill the cell by degrading proteins indiscriminately.
Research on apoptosis has increased substantially since the early 1990s. In addition to its importance as a biological phenomenon, defective apoptotic processes have been implicated in a wide variety of diseases. Excessive apoptosis causes atrophy, whereas an insufficient amount results in uncontrolled cell proliferation, such as cancer. Some factors like Fas receptors and caspases promote apoptosis, while some members of the Bcl-2 family of proteins inhibit apoptosis.
Discovery and etymology
German scientist Karl Vogt was first to describe the principle of apoptosis in 1842. In 1885, anatomist Walther Flemming delivered a more precise description of the process of programmed cell death. However, it was not until 1965 that the topic was resurrected. While studying tissues using electron microscopy, John Foxton Ross Kerr at University of Queensland was able to distinguish apoptosis from traumatic cell death.[6] Following the publication of a paper describing the phenomenon, Kerr was invited to join Alastair R Currie, as well as Andrew Wyllie, who was Currie's graduate student,[7] at University of Aberdeen. In 1972, the trio published a seminal article in the British Journal of Cancer.[8] Kerr had initially used the term programmed cell necrosis, but in the article, the process of natural cell death was called apoptosis. Kerr, Wyllie and Currie credited James Cormack, a professor of Greek language at University of Aberdeen, with suggesting the term apoptosis. Kerr received the Paul Ehrlich and Ludwig Darmstaedter Prize on March 14, 2000, for his description of apoptosis. He shared the prize with Boston biologist H. Robert Horvitz.[9]
For many years, the terms "apoptosis" and "programmed cell death" were not highly cited. What transformed cell death from obscurity to a major field of research were two things: the identification of components of the cell death control and effector mechanisms, and the linkage of abnormalities in cell death to human disease, in particular cancer.
The 2002 Nobel Prize in Medicine was awarded to Sydney Brenner, Horvitz and John E. Sulston for their work identifying genes that control apoptosis. The genes were identified by studies in the nematode C. elegans and these same genes function in humans for apoptosis.

In Greek, apoptosis translates to the "falling off" of leaves from a tree.[10] Cormack, professor of Greek language, reintroduced the term for medical use as it had a medical meaning for the Greeks over two thousand years before. Hippocrates used the term to mean "the falling off of the bones". Galen extended its meaning to "the dropping of the scabs". Cormack was no doubt aware of this usage when he suggested the name. Debate continues over the correct pronunciation, with opinion divided between a pronunciation with the second p silent (/æpəˈtoʊsᵻs/ ap-ə-TOH-sis[11][12]) and the second p pronounced (/æpəpˈtoʊsᵻs/),[11][13] as in the original Greek. In English, the p of the Greek -pt- consonant cluster is typically silent at the beginning of a word (e.g. pterodactyl, Ptolemy), but articulated when used in combining forms preceded by a vowel, as in helicopter or the orders of insects: diptera, lepidoptera, etc.
In the original Kerr, Wyllie & Currie paper,[8] there is a footnote regarding the pronunciation:
"We are most grateful to Professor James Cormack of the Department of Greek, University of Aberdeen, for suggesting this term. The word "apoptosis" (ἀπόπτωσις) is used in Greek to describe the "dropping off" or "falling off" of petals from flowers, or leaves from trees. To show the derivation clearly, we propose that the stress should be on the penultimate syllable, the second half of the word being pronounced like "ptosis" (with the "p" silent), which comes from the same root "to fall", and is already used to describe the drooping of the upper eyelid."
Activation mechanisms
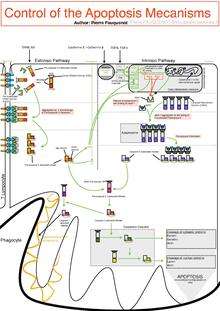
The initiation of apoptosis is tightly regulated by activation mechanisms, because once apoptosis has begun, it inevitably leads to the death of the cell.[14] The two best-understood activation mechanisms are the intrinsic pathway (also called the mitochondrial pathway) and the extrinsic pathway.[15] The intrinsic pathway is activated by intracellular signals generated when cells are stressed and depends on the release of proteins from the intermembrane space of mitochondria.[16] The extrinsic pathway is activated by extracellular ligands binding to cell-surface death receptors, which leads to the formation of the death-inducing signaling complex (DISC).[17]
A cell initiates intracellular apoptotic signaling in response to a stress, which may bring about cell suicide. The binding of nuclear receptors by glucocorticoids,[18] heat,[18] radiation,[18] nutrient deprivation,[18] viral infection,[18] hypoxia[18] and increased intracellular calcium concentration,[19][20] for example, by damage to the membrane, can all trigger the release of intracellular apoptotic signals by a damaged cell. A number of cellular components, such as poly ADP ribose polymerase, may also help regulate apoptosis.[21]
Before the actual process of cell death is precipitated by enzymes, apoptotic signals must cause regulatory proteins to initiate the apoptosis pathway. This step allows those signals to cause cell death, or the process to be stopped, should the cell no longer need to die. Several proteins are involved, but two main methods of regulation have been identified: the targeting of mitochondria functionality,[22] or directly transducing the signal via adaptor proteins to the apoptotic mechanisms. An extrinsic pathway for initiation identified in several toxin studies is an increase in calcium concentration within a cell caused by drug activity, which also can cause apoptosis via a calcium binding protease calpain.
Intrinsic pathway
The mitochondria are essential to multicellular life. Without them, a cell ceases to respire aerobically and quickly dies. This fact forms the basis for some apoptotic pathways. Apoptotic proteins that target mitochondria affect them in different ways. They may cause mitochondrial swelling through the formation of membrane pores, or they may increase the permeability of the mitochondrial membrane and cause apoptotic effectors to leak out.[18][23] They are very closely related to intrinsic pathway, and tumors arise more frequently through intrinsic pathway than the extrinsic pathway because of sensitivity.[24] There is also a growing body of evidence indicating that nitric oxide is able to induce apoptosis by helping to dissipate the membrane potential of mitochondria and therefore make it more permeable.[25] Nitric oxide has been implicated in initiating and inhibiting apoptosis through its possible action as a signal molecule of subsequent pathways that activate apoptosis.[26]
Mitochondrial proteins known as SMACs (second mitochondria-derived activator of caspases) are released into the cell's cytosol following the increase in permeability of the mitochondia membranes. SMAC binds to proteins that inhibit apoptosis (IAPs) thereby deactivating them, and preventing the IAPs from arresting the process and therefore allowing apoptosis to proceed. IAP also normally suppresses the activity of a group of cysteine proteases called caspases,[27] which carry out the degradation of the cell. Therefore, the actual degradation enzymes can be seen to be indirectly regulated by mitochondrial permeability.
Cytochrome c is also released from mitochondria due to formation of a channel, the mitochondrial apoptosis-induced channel (MAC), in the outer mitochondrial membrane,[28] and serves a regulatory function as it precedes morphological change associated with apoptosis.[18] Once cytochrome c is released it binds with Apoptotic protease activating factor – 1 (Apaf-1) and ATP, which then bind to pro-caspase-9 to create a protein complex known as an apoptosome. The apoptosome cleaves the pro-caspase to its active form of caspase-9, which in turn activates the effector caspase-3.
MAC (not to be confused with the Membrane Attack Complex formed by complement activation, also commonly denoted as MAC), also called "Mitochondrial Outer Membrane Permeabilization Pore" is regulated by various proteins, such as those encoded by the mammalian Bcl-2 family of anti-apoptopic genes, the homologs of the ced-9 gene found in C. elegans.[29][30] Bcl-2 proteins are able to promote or inhibit apoptosis by direct action on MAC/MOMPP. Bax and/or Bak form the pore, while Bcl-2, Bcl-xL or Mcl-1 inhibit its formation.
Extrinsic pathway
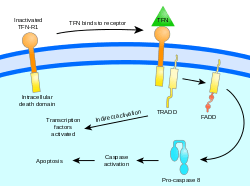
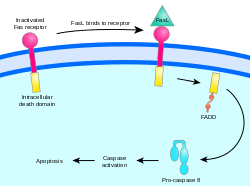
Two theories of the direct initiation of apoptotic mechanisms in mammals have been suggested: the TNF-induced (tumour necrosis factor) model and the Fas-Fas ligand-mediated model, both involving receptors of the TNF receptor (TNFR) family[31] coupled to extrinsic signals.
TNF path
TNF-alpha is a cytokine produced mainly by activated macrophages, and is the major extrinsic mediator of apoptosis. Most cells in the human body have two receptors for TNF-alpha: TNFR1 and TNFR2. The binding of TNF-alpha to TNFR1 has been shown to initiate the pathway that leads to caspase activation via the intermediate membrane proteins TNF receptor-associated death domain (TRADD) and Fas-associated death domain protein (FADD). cIAP1/2 can inhibit TNF-α signaling by binding to TRAF2. FLIP inhibits the activation of caspase-8.[32] Binding of this receptor can also indirectly lead to the activation of transcription factors involved in cell survival and inflammatory responses.[33] However, signalling through TNFR1 might also induce apoptosis in a caspase-independent manner.[34] The link between TNF-alpha and apoptosis shows why an abnormal production of TNF-alpha plays a fundamental role in several human diseases, especially in autoimmune diseases.
Fas path
The fas receptor First apoptosis signal (fas) – (also known as Apo-1 or CD95) binds the Fas ligand (FasL), a transmembrane protein part of the TNF family.[31] The interaction between Fas and FasL results in the formation of the death-inducing signaling complex (DISC), which contains the FADD, caspase-8 and caspase-10. In some types of cells (type I), processed caspase-8 directly activates other members of the caspase family, and triggers the execution of apoptosis of the cell. In other types of cells (type II), the Fas-DISC starts a feedback loop that spirals into increasing release of proapoptotic factors from mitochondria and the amplified activation of caspase-8.[35]
Common components
Following TNF-R1 and Fas activation in mammalian cells a balance between proapoptotic (BAX,[36] BID, BAK, or BAD) and anti-apoptotic (Bcl-Xl and Bcl-2) members of the Bcl-2 family is established. This balance is the proportion of proapoptotic homodimers that form in the outer-membrane of the mitochondrion. The proapoptotic homodimers are required to make the mitochondrial membrane permeable for the release of caspase activators such as cytochrome c and SMAC. Control of proapoptotic proteins under normal cell conditions of nonapoptotic cells is incompletely understood, but in general, Bax or Bak are activated by the activation of BH3-only proteins, part of the Bcl-2 family.
Caspases Caspases play the central role in the transduction of ER apoptotic signals. Caspases are proteins that are highly conserved, cysteine-dependent aspartate-specific proteases. There are two types of caspases: initiator caspases, caspase 2,8,9,10,11,12, and effector caspases, caspase 3,6,7. The activation of initiator caspases requires binding to specific oligomeric activator protein. Effector caspases are then activated by these active initiator caspases through proteolytic cleavage. The active effector caspases then proteolytically degrade a host of intracellular proteins to carry out the cell death program.
Caspase-independent apoptotic pathway There also exists a caspase-independent apoptotic pathway that is mediated by AIF (apoptosis-inducing factor).[37]
Apoptosis model in amphibians

Amphibian frog Xenopus laevis serves as an ideal model system for the study of the mechanisms of apoptosis. In fact, iodine and thyroxine also stimulate the spectacular apoptosis of the cells of the larval gills, tail and fins in amphibians metamorphosis, and stimulate the evolution of their nervous system transforming the aquatic, vegetarian tadpole into the terrestrial, carnivorous frog.[38][39][40][41]
Proteolytic caspase cascade: Killing the cell
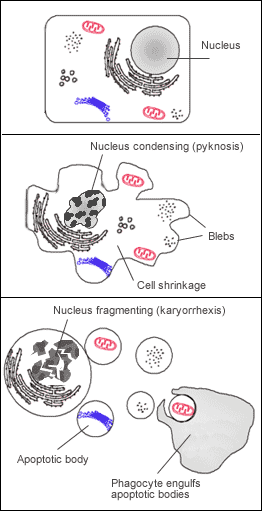
Many pathways and signals lead to apoptosis, but these converge on a single mechanism that actually causes the death of the cell. After a cell receives stimulus, it undergoes organized degradation of cellular organelles by activated proteolytic caspases. In addition to the destruction of cellular organelles, mRNA is rapidly and globally degraded by a mechanism that is not yet fully characterized.[42] mRNA decay is triggered very early in apoptosis. A cell undergoing apoptosis shows a characteristic morphology:
- Cell shrinkage and rounding are shown because of the breakdown of the proteinaceous cytoskeleton by caspases.[43]
- The cytoplasm appears dense, and the organelles appear tightly packed.
- Chromatin undergoes condensation into compact patches against the nuclear envelope (also known as the perinuclear envelope) in a process known as pyknosis, a hallmark of apoptosis.[44][45]
- The nuclear envelope becomes discontinuous and the DNA inside it is fragmented in a process referred to as karyorrhexis. The nucleus breaks into several discrete chromatin bodies or nucleosomal units due to the degradation of DNA.[46]
- The cell membrane shows irregular buds known as blebs.
- The cell breaks apart into multiple vesicles called apoptotic bodies, which are then phagocytosed.
Apoptosis progresses quickly and its products are quickly removed, making it difficult to detect or visualize. During karyorrhexis, endonuclease activation leaves short DNA fragments, regularly spaced in size. These give a characteristic "laddered" appearance on agar gel after electrophoresis. Tests for DNA laddering differentiate apoptosis from ischemic or toxic cell death.[47]
Removal of dead cells
The removal of dead cells by neighboring phagocytic cells has been termed efferocytosis.[48] Dying cells that undergo the final stages of apoptosis display phagocytotic molecules, such as phosphatidylserine, on their cell surface.[49] Phosphatidylserine is normally found on the inner leaflet surface of the plasma membrane, but is redistributed during apoptosis to the extracellular surface by a protein known as scramblase.[50] These molecules mark the cell for phagocytosis by cells possessing the appropriate receptors, such as macrophages.[51] The removal of dying cells by phagocytes occurs in an orderly manner without eliciting an inflammatory response.[52]
Pathway knock-outs
Many knock-outs have been made in the apoptosis pathways to test the function of each of the proteins. Several caspases, in addition to APAF-1 and FADD, have been mutated to determine the new phenotype. In order to create a tumor necrosis factor (TNF) knockout, an exon containing the nucleotides 3704-5364 was removed from the gene. This exon encodes a portion of the mature TNF domain, as well as the leader sequence, which is a highly conserved region necessary for proper intracellular processing. TNF-/- mice develop normally and have no gross structural or morphological abnormalities. However, upon immunization with SRBC (sheep red blood cells), these mice demonstrated a deficiency in the maturation of an antibody response; they were able to generate normal levels of IgM, but could not develop specific IgG levels. Apaf-1 is the protein that turns on caspase 9 by cleavage to begin the caspase cascade that leads to apoptosis. Since a -/- mutation in the APAF-1 gene is embryonic lethal, a gene trap strategy was used in order to generate an APAF-1 -/- mouse. This assay is used to disrupt gene function by creating an intragenic gene fusion. When an APAF-1 gene trap is introduced into cells, many morphological changes occur, such as spina bifida, the persistence of interdigital webs, and open brain. In addition, after embryonic day 12.5, the brain of the embryos showed several structural changes. APAF-1 cells are protected from apoptosis stimuli such as irradiation. A BAX-1 knock-out mouse exhibits normal forebrain formation and a decreased programmed cell death in some neuronal populations and in the spinal cord, leading to an increase in motor neurons.
The caspase proteins are integral parts of the apoptosis pathway, so it follows that knock-outs made have varying damaging results. A caspase 9 knock-out leads to a severe brain malformation. A caspase 8 knock-out leads to cardiac failure and thus embryonic lethality. However, with the use of cre-lox technology, a caspase 8 knock-out has been created that exhibits an increase in peripheral T cells, an impaired T cell response, and a defect in neural tube closure. These mice were found to be resistant to apoptosis mediated by CD95, TNFR, etc. but not resistant to apoptosis caused by UV irradiation, chemotherapeutic drugs, and other stimuli. Finally, a caspase 3 knock-out was characterized by ectopic cell masses in the brain and abnormal apoptotic features such as membrane blebbing or nuclear fragmentation. A remarkable feature of these KO mice is that they have a very restricted phenotype: Casp3, 9, APAF-1 KO mice have deformations of neural tissue and FADD and Casp 8 KO showed defective heart development, however in both types of KO other organs developed normally and some cell types were still sensitive to apoptotic stimuli suggesting that unknown proapoptotic pathways exist.
Methods for distinguishing apoptotic from necrotic (necroptotic) cells
In order to perform analysis of apoptotic versus necrotic (necroptotic) cells, one can do analysis of morphology by time-lapse microscopy, flow fluorocytometry, and transmission electron microscopy. There are also various biochemical techniques for analysis of cell surface markers (phosphatidylserine exposure versus cell permeability by flow fluorocytometry), cellular markers such as DNA fragmentation[53] (flow fluorocytometry), caspase activation, Bid cleavage, and cytochrome c release (Western blotting). It is important to know how primary and secondary necrotic cells can be distinguished by analysis of supernatant for caspases, HMGB1, and release of cytokeratin 18. However, no distinct surface or biochemical markers of necrotic cell death have been identified yet, and only negative markers are available. These include absence of apoptotic markers (caspase activation, cytochrome c release, and oligonucleosomal DNA fragmentation) and differential kinetics of cell death markers (phosphatidylserine exposure and cell membrane permeabilization). A selection of techniques that can be used to distinguish apoptosis from necroptotic cells could be found in these references.[54][55][56]
Implication in disease
Defective pathways
The many different types of apoptotic pathways contain a multitude of different biochemical components, many of them not yet understood.[57] As a pathway is more or less sequential in nature, removing or modifying one component leads to an effect in another. In a living organism, this can have disastrous effects, often in the form of disease or disorder. A discussion of every disease caused by modification of the various apoptotic pathways would be impractical, but the concept overlying each one is the same: The normal functioning of the pathway has been disrupted in such a way as to impair the ability of the cell to undergo normal apoptosis. This results in a cell that lives past its "use-by-date" and is able to replicate and pass on any faulty machinery to its progeny, increasing the likelihood of the cell's becoming cancerous or diseased.
A recently described example of this concept in action can be seen in the development of a lung cancer called NCI-H460.[58] The X-linked inhibitor of apoptosis protein (XIAP) is overexpressed in cells of the H460 cell line. XIAPs bind to the processed form of caspase-9, and suppress the activity of apoptotic activator cytochrome c, therefore overexpression leads to a decrease in the amount of proapoptotic agonists. As a consequence, the balance of anti-apoptotic and proapoptotic effectors is upset in favour of the former, and the damaged cells continue to replicate despite being directed to die.
Dysregulation of p53
The tumor-suppressor protein p53 accumulates when DNA is damaged due to a chain of biochemical factors. Part of this pathway includes alpha-interferon and beta-interferon, which induce transcription of the p53 gene, resulting in the increase of p53 protein level and enhancement of cancer cell-apoptosis.[59] p53 prevents the cell from replicating by stopping the cell cycle at G1, or interphase, to give the cell time to repair, however it will induce apoptosis if damage is extensive and repair efforts fail. Any disruption to the regulation of the p53 or interferon genes will result in impaired apoptosis and the possible formation of tumors.
Inhibition
Inhibition of apoptosis can result in a number of cancers, autoimmune diseases, inflammatory diseases, and viral infections. It was originally believed that the associated accumulation of cells was due to an increase in cellular proliferation, but it is now known that it is also due to a decrease in cell death. The most common of these diseases is cancer, the disease of excessive cellular proliferation, which is often characterized by an overexpression of IAP family members. As a result, the malignant cells experience an abnormal response to apoptosis induction: Cycle-regulating genes (such as p53, ras or c-myc) are mutated or inactivated in diseased cells, and further genes (such as bcl-2) also modify their expression in tumors.
HeLa cell
Apoptosis in HeLa cells is inhibited by proteins produced by the cell; these inhibitory proteins target retinoblastoma tumor-suppressing proteins.[60] These tumor-suppressing proteins regulate the cell cycle, but are rendered inactive when bound to an inhibitory protein.[60] HPV E6 and E7 are inhibitory proteins expressed by the human papillomavirus, HPV being responsible for the formation of the cervical tumor from which HeLa cells are derived.[61] HPV E6 causes p53, which regulates the cell cycle, to become inactive.[62] HPV E7 binds to retinoblastoma tumor suppressing proteins and limits its ability to control cell division.[62] These two inhibitory proteins are partially responsible for HeLa cells' immortality by inhibiting apoptosis to occur.[63] CDV (Canine Distemper Virus) is able to induce apoptosis despite the presence of these inhibitory proteins. This is an important oncolytic property of CDV: this virus is capable of killing canine lymphoma cells. Oncoproteins E6 and E7 still leave p53 inactive, but they are not able to avoid the activation of caspases induced from the stress of viral infection. These oncolytic properties provided a promising link between CDV and lymphoma apoptosis, which can lead to development of alternative treatment methods for both canine lymphoma and human non-Hodgkin lymphoma. Defects in the cell cycle are thought to be responsible for the resistance to chemotherapy or radiation by certain tumor cells, so a virus that can induce apoptosis despite defects in the cell cycle is useful for cancer treatment.[63]
Treatments
The main method of treatment for death from signaling-related diseases involves either increasing or decreasing the susceptibility of apoptosis in diseased cells, depending on whether the disease is caused by either the inhibition of or excess apoptosis. For instance, treatments aim to restore apoptosis to treat diseases with deficient cell death, and to increase the apoptotic threshold to treat diseases involved with excessive cell death. To stimulate apoptosis, one can increase the number of death receptor ligands (such as TNF or TRAIL), antagonize the anti-apoptotic Bcl-2 pathway, or introduce Smac mimetics to inhibit the inhibitor (IAPs). The addition of agents such as Herceptin, Iressa, or Gleevec works to stop cells from cycling and causes apoptosis activation by blocking growth and survival signaling further upstream. Finally, adding p53-MDM2 complexes displaces p53 and activates the p53 pathway, leading to cell cycle arrest and apoptosis. Many different methods can be used either to stimulate or to inhibit apoptosis in various places along the death signaling pathway.[64]
Apoptosis is a multi-step, multi-pathway cell-death programme that is inherent in every cell of the body. In cancer, the apoptosis cell-division ratio is altered. Cancer treatment by chemotherapy and irradiation kills target cells primarily by inducing apoptosis.
Hyperactive apoptosis
On the other hand, loss of control of cell death (resulting in excess apoptosis) can lead to neurodegenerative diseases, hematologic diseases, and tissue damage. The progression of HIV is directly linked to excess, unregulated apoptosis. In a healthy individual, the number of CD4+ lymphocytes is in balance with the cells generated by the bone marrow; however, in HIV-positive patients, this balance is lost due to an inability of the bone marrow to regenerate CD4+ cells. In the case of HIV, CD4+ lymphocytes die at an accelerated rate through uncontrolled apoptosis, when stimulated.
Treatments
Treatments aiming to inhibit works to block specific caspases. Finally, the Akt protein kinase promotes cell survival through two pathways. Akt phosphorylates and inhibits Bas (a Bcl-2 family member), causing Bas to interact with the 14-3-3 scaffold, resulting in Bcl dissociation and thus cell survival. Akt also activates IKKα, which leads to NF-κB activation and cell survival. Active NF-κB induces the expression of anti-apoptotic genes such as Bcl-2, resulting in inhibition of apoptosis. NF-κB has been found to play both an antiapoptotic role and a proapoptotic role depending on the stimuli utilized and the cell type.[65]
HIV progression
The progression of the human immunodeficiency virus infection into AIDS is due primarily to the depletion of CD4+ T-helper lymphocytes in a manner that is too rapid for the body's bone marrow to replenish the cells, leading to a compromised immune system. One of the mechanisms by which T-helper cells are depleted is apoptosis, which results from a series of biochemical pathways:[66]
- HIV enzymes deactivate anti-apoptotic Bcl-2. This does not directly cause cell death but primes the cell for apoptosis should the appropriate signal be received. In parallel, these enzymes activate proapoptotic procaspase-8, which does directly activate the mitochondrial events of apoptosis.
- HIV may increase the level of cellular proteins that prompt Fas-mediated apoptosis.
- HIV proteins decrease the amount of CD4 glycoprotein marker present on the cell membrane.
- Released viral particles and proteins present in extracellular fluid are able to induce apoptosis in nearby "bystander" T helper cells.
- HIV decreases the production of molecules involved in marking the cell for apoptosis, giving the virus time to replicate and continue releasing apoptotic agents and virions into the surrounding tissue.
- The infected CD4+ cell may also receive the death signal from a cytotoxic T cell.
Cells may also die as direct consequences of viral infections. HIV-1 expression induces tubular cell G2/M arrest and apoptosis.[67] The progression from HIV to AIDS is not immediate or even necessarily rapid; HIV's cytotoxic activity toward CD4+ lymphocytes is classified as AIDS once a given patient's CD4+ cell count falls below 200.[68]
Viral infection
Viral induction of apoptosis occurs when one or several cells of a living organism are infected with a virus, leading to cell death. Cell death in organisms is necessary for the normal development of cells and the cell cycle maturation.[69] It is also important in maintaining the regular functions and activities of cells.
Viruses can trigger apoptosis of infected cells via a range of mechanisms including:
- Receptor binding
- Activation of protein kinase R (PKR)
- Interaction with p53
- Expression of viral proteins coupled to MHC proteins on the surface of the infected cell, allowing recognition by cells of the immune system (such as Natural Killer and cytotoxic T cells) that then induce the infected cell to undergo apoptosis.[70]
Canine distemper virus (CDV) is known to cause apoptosis in central nervous system and lymphoid tissue of infected dogs in vivo and in vitro.[71] Apoptosis caused by CDV is typically induced via the extrinsic pathway, which activates caspases that disrupt cellular function and eventually leads to the cells death.[60] In normal cells, CDV activates caspase-8 first, which works as the initiator protein followed by the executioner protein caspase-3.[60] However, apoptosis induced by CDV in HeLa cells does not involve the initiator protein caspase-8. HeLa cell apoptosis caused by CDV follows a different mechanism than that in vero cell lines.[60] This change in the caspase cascade suggests CDV induces apoptosis via the intrinsic pathway, excluding the need for the initiator caspase-8. The executioner protein is instead activated by the internal stimuli caused by viral infection not a caspase cascade.[60]
The Oropouche virus (OROV) is found in the family Bunyaviridae. The study of apoptosis brought on by Bunyaviridae was initiated in 1996, when it was observed that apoptosis was induced by the La Crosse virus into the kidney cells of baby hamsters and into the brains of baby mice.[72]
OROV is a disease that is transmitted between humans by the biting midge (Culicoides paraensis).[73] It is referred to as a zoonotic arbovirus and causes febrile illness, characterized by the onset of a sudden fever known as Oropouche fever.[74]
The Oropouche virus also causes disruption in cultured cells – cells that are cultivated in distinct and specific conditions. An example of this can be seen in HeLa cells, whereby the cells begin to degenerate shortly after they are infected.[72]
With the use of gel electrophoresis, it can be observed that OROV causes DNA fragmentation in HeLa cells. It can be interpreted by counting, measuring, and analyzing the cells of the Sub/G1 cell population.[72] When HeLA cells are infected with OROV, the cytochrome C is released from the membrane of the mitochondria, into the cytosol of the cells. This type of interaction shows that apoptosis is activated via an intrinsic pathway.[69]
In order for apoptosis to occur within OROV, viral uncoating, viral internalization, along with the replication of cells is necessary. Apoptosis in some viruses is activated by extracellular stimuli. However, studies have demonstrated that the OROV infection causes apoptosis to be activated through intracellular stimuli and involves the mitochondria.[72]
Many viruses encode proteins that can inhibit apoptosis.[75] Several viruses encode viral homologs of Bcl-2. These homologs can inhibit proapoptotic proteins such as BAX and BAK, which are essential for the activation of apoptosis. Examples of viral Bcl-2 proteins include the Epstein-Barr virus BHRF1 protein and the adenovirus E1B 19K protein.[76] Some viruses express caspase inhibitors that inhibit caspase activity and an example is the CrmA protein of cowpox viruses. Whilst a number of viruses can block the effects of TNF and Fas. For example, the M-T2 protein of myxoma viruses can bind TNF preventing it from binding the TNF receptor and inducing a response.[77] Furthermore, many viruses express p53 inhibitors that can bind p53 and inhibit its transcriptional transactivation activity. As a consequence, p53 cannot induce apoptosis, since it cannot induce the expression of proapoptotic proteins. The adenovirus E1B-55K protein and the hepatitis B virus HBx protein are examples of viral proteins that can perform such a function.[78]
Viruses can remain intact from apoptosis in particular in the latter stages of infection. They can be exported in the apoptotic bodies that pinch off from the surface of the dying cell, and the fact that they are engulfed by phagocytes prevents the initiation of a host response. This favours the spread of the virus.[77]
Plants
Programmed cell death in plants has a number of molecular similarities to that of animal apoptosis, but it also has differences, notable ones being the presence of a cell wall and the lack of an immune system that removes the pieces of the dead cell. Instead of an immune response, the dying cell synthesizes substances to break itself down and places them in a vacuole that ruptures as the cell dies. Whether this whole process resembles animal apoptosis closely enough to warrant using the name apoptosis (as opposed to the more general programmed cell death) is unclear.[79]
Caspase-independent apoptosis
The characterization of the caspases allowed the development of caspase inhibitors, which can be used to determine whether a cellular process involves active caspases. Using these inhibitors it was discovered that cells can die while displaying a morphology similar to apoptosis without caspase activation.[80] Later studies linked this phenomenon to the release of AIF (apoptosis-inducing factor) from the mitochondria and its translocation into the nucleus mediated by its NLS (nuclear localization signal). Inside the mitochondria, AIF is anchored to the inner membrane. In order to be released, the protein is cleaved by a calcium-dependent calpain protease.
Apoptosis protein subcellular location prediction
In 2003, a method was developed for predicting subcellular location of apoptosis proteins.[81] Subsequent to this, various modes of Chou's pseudo amino acid composition were developed for improving the quality of predicting subcellular localization of apoptosis proteins based on their sequence information alone.[82][83][84][85]
See also
Footnotes
References
- ↑ "Quantitative phase contrast microscopy — label-free live cell imaging and quantification". Phase Holographic Imaging AB.
- ↑ Green, Douglas (2011). Means to an End: Apoptosis and other Cell Death Mechanisms. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. ISBN 978-0-87969-888-1.
- ↑ Alberts, p. 2.
- ↑ Karam, Jose A. (2009). Apoptosis in Carcinogenesis and Chemotherapy. Netherlands: Springer. ISBN 978-1-4020-9597-9.
- ↑ Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2008). "Chapter 18 Apoptosis: Programmed Cell Death Eliminates Unwanted Cells". Molecular Biology of the Cell (textbook) (5th ed.). Garland Science. p. 1115. ISBN 978-0-8153-4105-5.
- ↑ Kerr, JF. (1965). "A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes". Journal of Pathology and Bacteriology. 90 (2): 419–35. doi:10.1002/path.1700900210.
- ↑ Agency for Science, Technology and Research. "Prof Andrew H. Wyllie – Lecture Abstract". Archived from the original on 2007-11-13. Retrieved 2007-03-30.
- 1 2 Kerr JF, Wyllie AH, Currie AR (August 1972). "Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics". Br. J. Cancer. 26 (4): 239–57. doi:10.1038/bjc.1972.33. PMC 2008650
 . PMID 4561027.
. PMID 4561027. - ↑ O'Rourke MG, Ellem KA (2000). "John Kerr and apoptosis". Med. J. Aust. 173 (11–12): 616–7. PMID 11379508.
- ↑ Alberts, p. 1021.
- 1 2 US dict: ăp′·ə·tō′·sĭs (American Heritage Dictionary Archived June 30, 2008, at the Wayback Machine.)
- ↑ Apoptosis Interest Group (1999). "About apoptosis". Archived from the original on 28 December 2006. Retrieved 2006-12-15.
- ↑ Webster.com dictionary entry
- ↑ Alberts, p. 1029.
- ↑ Alberts, p. 1023.
- ↑ Alberts, p. 1032.
- ↑ Alberts, p. 1024.
- 1 2 3 4 5 6 7 8 Cotran, Ramzi, S.; Kumar, Collins (1998). Robbins Pathologic Basis of Disease. Philadelphia: W.B Saunders Company. ISBN 0-7216-7335-X.
- ↑ Mattson MP, Chan SL (2003). "Calcium orchestrates apoptosis". Nature Cell Biology. 5 (12): 1041–1043. doi:10.1038/ncb1203-1041. PMID 14647298.
- ↑ Uguz AC, Naziroglu M, Espino J, Bejarano I, González D, Rodríguez AB, Pariente JA (November 2009). "Selenium Modulates Oxidative Stress-Induced Cell Apoptosis in Human Myeloid HL-60 Cells Through Regulationof Calcium Release and Caspase-3 and -9 Activities". J Membrane Biol 232: 15-23. doi:[10.1007/s00232-009-9212-2].
- ↑ Chiarugi A, Moskowitz MA (2002). "PARP-1—a perpetrator of apoptotic cell death?". Science. 297 (5579): 259–63. doi:10.1126/science.1074592. PMID 12114611.
- ↑ Bejarano I, Espino J, González-Flores D, Casado JG, Redondo PC, Rosado JA, Barriga C, Pariente JA, Rodríguez AB (2009). "Role of Calcium Signals on Hydrogen Peroxide-Induced Apoptosis in Human Myeloid HL-60 Cells". International Journal of Biomedical Science 5(3): 246-256.
- ↑ D. González, I. Bejarano, C. Barriga, A.B. Rodríguez, J.A. Pariente (2010). "Oxidative Stress-Induced Caspases are Regulated in Human Myeloid HL-60 Cells by Calcium Signal". Current Signal Transduction Therapy 5: 181-186. doi:[10.2174/157436210791112172]
- ↑ Mohan S, Bustamam A, Abdelwahab SI, Al-Zubairi AS, Aspollah M, Abdullah R, Elhassan MM, Ibrahim MY, Syam S: Typhonium flagelliforme induces apoptosis in CEMss cells via activation of caspase-9, PARP cleavage and cytochrome c release: Its activation coupled with G0/G1 phase cell cycle arrest. Journal of Ethnopharmacology 2010
- ↑ Brüne B (August 2003). "Nitric oxide: NO apoptosis or turning it ON?". Cell Death Differ. 10 (8): 864–9. doi:10.1038/sj.cdd.4401261. PMID 12867993.
- ↑ Brune, B. et al. 1999. "Nitric oxide (NO): an effector of apoptosis." Cell Death and Differentiation. 6:10 pg 969-975.
- ↑ Fesik SW, Shi Y (2001). "Controlling the caspases". Science. 294 (5546): 1477–8. doi:10.1126/science.1062236. PMID 11711663.
- ↑ Laurent M. Dejean; Sonia Martinez-Caballero; Kathleen W. Kinnally (2006). "Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis?". Cell Death and Differentiation. 13 (8): 1387–5. doi:10.1038/sj.cdd.4401949. PMID 16676005.
- ↑ Dejean LM, Martinez-Caballero S, Manon S, Kinnally KW (February 2006). "Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins". Biochim. Biophys. Acta. 1762 (2): 191–201. doi:10.1016/j.bbadis.2005.07.002. PMID 16055309.
- ↑ Lodish, Harvey; et al. (2004). Molecular Cell Biology. New York: W.H. Freedman and Company. ISBN 0-7167-4366-3.
- 1 2 Wajant H (2002). "The Fas signaling pathway: more than a paradigm". Science. 296 (5573): 1635–6. doi:10.1126/science.1071553. PMID 12040174.
- ↑ Chen G, Goeddel DV (2002). "TNF-R1 signaling: a beautiful pathway". Science. 296 (5573): 1634–5. doi:10.1126/science.1071924. PMID 12040173.
- ↑ Goeddel, DV. "Connection Map for Tumor Necrosis Factor Pathway". Science STKE. 2007: tw132. doi:10.1126/stke.3822007tw132.
- ↑ Chen W, Li N, Chen T, Han Y, Li C, Wang Y, et al. (2005). "The lysosome-associated apoptosis-inducing protein containing the pleckstrin homology (PH) and FYVE domains (LAPF), representative of a novel family of PH and FYVE domain-containing proteins, induces caspase-independent apoptosis via the lysosomal-mitochondrial pathway.". Journal of Biological Chemistry. 280 (49): 40985–40995. doi:10.1074/jbc.M502190200. PMID 16188880.
- ↑ Wajant, H. "Connection Map for Fas Signaling Pathway". Science STKE. 2007: tr1. doi:10.1126/stke.3802007tr1.
- ↑ Murphy KM, Ranganathan V, Farnsworth ML, Kavallaris M, Lock RB (January 2000). "Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells". Cell Death Differ. 7 (1): 102–11. doi:10.1038/sj.cdd.4400597. PMID 10713725.
- ↑ Susin SA, Lorenzo HK, Zamzami N (February 1999). "Molecular characterization of mitochondrial apoptosis-inducing factor". Nature. 397 (6718): 441–6. doi:10.1038/17135. PMID 9989411.
- ↑ Jewhurst K, Levin M, McLaughlin KA (2014). "Optogenetic Control of Apoptosis in Targeted Tissues of Xenopus laevis Embryos". J Cell Death. 7: 25–31. doi:10.4137/JCD.S18368. PMC 4213186. PMID 25374461.
- ↑ Venturi, Sebastiano (2011). "Evolutionary Significance of Iodine". Current Chemical Biology-. 5 (3): 155–162. doi:10.2174/187231311796765012. ISSN 1872-3136.
- ↑ Venturi, Sebastiano (2014). "Iodine, PUFAs and Iodolipids in Health and Disease: An Evolutionary Perspective". Human Evolution-. 29 (1-3): 185–205. ISSN 0393-9375.
- ↑ Tamura K, Takayama S, Ishii T, Mawaribuchi S, Takamatsu N, Ito M (2015). "Apoptosis and differentiation of Xenopus tail-derived myoblasts by thyroid hormone.". J Mol Endocrinol. 54 (3): 185–92. doi:10.1530/JME-14-0327.
- ↑ Thomas, M. P.; Liu, X; Whangbo, J; McCrossan, G; Sanborn, K. B.; Basar, E; Walch, M; Lieberman, J (2015). "Apoptosis Triggers Specific, Rapid, and Global mRNA Decay with 3' Uridylated Intermediates Degraded by DIS3L2". Cell Reports. 11: 1079–89. doi:10.1016/j.celrep.2015.04.026. PMID 25959823.
- ↑ Böhm I. Disruption of the cytoskeleton after apoptosis induction by autoantibodies. Autoimmunity 2003;36:183–189
- ↑ Susin, S; Daugas, E; Ravagnan, L; Samejima, K; Zamzami, N; Loeffler, M; Costantini, P; Ferri, KF; et al. (2000). "Two Distinct Pathways Leading to Nuclear Apoptosis". Journal of Experimental Medicine. 192 (4): 571–80. doi:10.1084/jem.192.4.571. PMC 2193229. PMID 10952727.
- ↑ Madeleine Kihlmark; Imreh, G; Hallberg, E (15 October 2001). "Sequential degradation of proteins from the nuclear envelope during apoptosis". Journal of Cell Science. 114 (20): 3643–53. PMID 11707516.
- ↑ Nagata S (April 2000). "Apoptotic DNA fragmentation". Exp. Cell Res. 256 (1): 12–8. doi:10.1006/excr.2000.4834. PMID 10739646.
- ↑ M Iwata; D Myerson; B Torok-Storb; RA Zager (1996). "An evaluation of renal tubular DNA laddering in response to oxygen deprivation and oxidant injury". Retrieved 2006-04-17.
- ↑ Vandivier RW, Henson PM, Douglas IS (June 2006). "Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease". Chest. 129 (6): 1673–82. doi:10.1378/chest.129.6.1673. PMID 16778289.
- ↑ Li MO; Sarkisian, MR; Mehal, WZ; Rakic, P; Flavell, RA (2003). "Phosphatidylserine receptor is required for clearance of apoptotic cells". Science. 302 (5650): 1560–3. doi:10.1126/science.1087621. PMID 14645847.
- ↑ Wang X; Wu, YC; Fadok, VA; Lee, MC; Gengyo-Ando, K; Cheng, LC; Ledwich, D; Hsu, PK; et al. (2003). "Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12". Science. 302 (5650): 1563–1566. doi:10.1126/science.1087641. PMID 14645848.
- ↑ Savill J, Gregory C, Haslett C. (2003). "Eat me or die". Science. 302 (5650): 1516–7. doi:10.1126/science.1092533. PMID 14645835.
- ↑ Krysko DV; Vandenabeele P. Phagocytosis of dying cells: from molecular mechanisms to human diseases. ISBN 978-1-4020-9292-3.
- ↑ Lozano G.M., Bejarano, I., Espino, J., González, D., Ortiz, A., García, J.F., Rodríguez, A.B., Pariente, J.A. (2009). "Density gradient capacitation is the most suitable method to improve fertilization and to reduce DNA fragmentation positive spermatozoa of infertile men". Anatolian Journal of Obstetrics & Gynecology 3(1): 1-7.
- ↑ Krysko DV, Vanden Berghe T, Parthoens E, D'Herde K, Vandenabeele P (2008). "Methods for distinguishing apoptotic from necrotic cells and measuring their clearance.". Methods Enzymol. 442: 307–41. doi:10.1016/S0076-6879(08)01416-X. PMID 18662577.
- ↑ Krysko DV, Vanden Berghe T, D'Herde K, Vandenabeele P (2008). "Apoptosis and necrosis: detection, discrimination and phagocytosis.". Methods. 44 (3): 205–21. doi:10.1016/j.ymeth.2007.12.001. PMID 18314051.
- ↑ Vanden Berghe T, Grootjans S, Goossens V, Dondelinger Y, Krysko DV, Takahashi N, Vandenabeele P (2013). "Determination of apoptotic and necrotic cell death in vitro and in vivo.". Methods. 61 (2): 117–29. doi:10.1016/j.ymeth.2013.02.011. PMID 23473780.
- ↑ Thompson, CB (1995). "Apoptosis in the pathogenesis and treatment of disease". Science. 267 (5203): 1456–62. doi:10.1126/science.7878464. PMID 7878464.
- ↑ Yang L, Mashima T, Sato S (February 2003). "Predominant suppression of apoptosome by inhibitor of apoptosis protein in non-small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine-conjugated Smac peptide". Cancer Res. 63 (4): 831–7. PMID 12591734.
- ↑ Takaoka A; Hayakawa, S; Yanai, H; Stoiber, D; Negishi, H; Kikuchi, H; Sasaki, S; Imai, K; et al. (2003). "Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence". Nature. 424 (6948): 516–23. doi:10.1038/nature01850. PMID 12872134.
- 1 2 3 4 5 6 Del Puerto HL, Martins AS, Milsted A, et al. (2011). "Canine distemper virus induces apoptosis in cervical tumor derived cell lines". Virol. J. 8 (1): 334. doi:10.1186/1743-422X-8-334. PMC 3141686. PMID 21718481.
- ↑ Liu HC, Chen GG, Vlantis AC, Tse GM, Chan AT, van Hasselt CA (March 2008). "Inhibition of apoptosis in human laryngeal cancer cells by E6 and E7 oncoproteins of human papillomavirus 16". J. Cell. Biochem. 103 (4): 1125–43. doi:10.1002/jcb.21490. PMID 17668439.
- 1 2 Niu XY, Peng ZL, Duan WQ, Wang H, Wang P (2006). "Inhibition of HPV 16 E6 oncogene expression by RNA interference in vitro and in vivo". Int. J. Gynecol. Cancer. 16 (2): 743–51. doi:10.1111/j.1525-1438.2006.00384.x. PMID 16681755.
- 1 2 Liu, Y; McKalip, A; Herman, B (2000). "Human Papillomavirus Type 16 E6 and HPV-16 E6/E7 Sensitize Human Keratinocytes to Apoptosis Induced by Chemotherapeutic Agents: Roles of p53 and Caspase Activation". Journal of Cellular Biochemistry. 78: 334–349. doi:10.1002/(sici)1097-4644(20000801)78:2<334::aid-jcb15>3.3.co;2-6.
- ↑ Boehm, I. (2006). "Apoptosis in physiological and pathological skin: Implications for therapy". Current Molecular Medicine. 6 (4): 375–394. doi:10.2174/156652406777435390. PMID 16900661.
- ↑ Farhana, Lulu. "Apoptosis Induction by a Novel Retinoid-Related Molecule Requires Nuclear Factor- κB Activation" (PDF). American Association of Cancer Research (AACR). Retrieved 2012-01-24.
- ↑ Judie B. Alimonti; T. Blake Ball; Keith R. Fowke (2003). "Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS". J. Gen. Virology. 84 (7): 1649–61. doi:10.1099/vir.0.19110-0. PMID 12810858.
- ↑ Vashistha H, Husain M, Kumar D, Yadav A, Arora S, Singhal PC. (2008) Ren Fail. 2008;30(6):655-64.
- ↑ Indiana University Health. "AIDS Defining Criteria | Riley". IU Health. Retrieved 2013-01-20.
- 1 2 Indran IR, Tufo G, Pervaiz S, Brenner C (June 2011). "Recent advances in apoptosis, mitochondria and drug resistance in cancer cells". Biochim. Biophys. Acta. 1807 (6): 735–45. doi:10.1016/j.bbabio.2011.03.010. PMID 21453675.
- ↑ Everett, H.; McFadden, G. (1999). "Apoptosis: an innate immune response to virus infection". Trends Microbiol. 7 (4): 160–5. doi:10.1016/S0966-842X(99)01487-0. PMID 10217831.
- ↑ Nishi T, Tsukiyama-Kohara K, Togashi K, Kohriyama N, Kai C (November 2004). "Involvement of apoptosis in syncytial cell death induced by canine distemper virus". Comp. Immunol. Microbiol. Infect. Dis. 27 (6): 445–55. doi:10.1016/j.cimid.2004.01.007. PMID 15325517.
- 1 2 3 4 Acrani GO, Gomes R, Proença-Módena JL, et al. (April 2010). "Apoptosis induced by Oropouche virus infection in HeLa cells is dependent on virus protein expression". Virus Res. 149 (1): 56–63. doi:10.1016/j.virusres.2009.12.013. PMID 20080135.
- ↑ Azevedo RS, Nunes MR, Chiang JO, et al. (June 2007). "Reemergence of Oropouche fever, northern Brazil". Emerging Infect. Dis. 13 (6): 912–5. doi:10.3201/eid1306.061114. PMC 2792853. PMID 17553235.
- ↑ Santos RI, Rodrigues AH, Silva ML, et al. (December 2008). "Oropouche virus entry into HeLa cells involves clathrin and requires endosomal acidification". Virus Res. 138 (1–2): 139–43. doi:10.1016/j.virusres.2008.08.016. PMID 18840482.
- ↑ Teodoro, J.G. Branton, P.E. (1997). "Regulation of apoptosis by viral gene products". J. Virol. 71 (3): 1739–46. PMC 191242. PMID 9032302.
- ↑ Polster, B.M. Pevsner, J. and Hardwick, J.M. (2004). "Viral Bcl-2 homologs and their role in virus replication and associated diseases". Biochim. Biophys. Acta. 1644 (2–3): 211–27. doi:10.1016/j.bbamcr.2003.11.001. PMID 14996505.
- 1 2 Hay, S.; Kannourakis, G. (2002). "A time to kill: viral manipulation of the cell death program". J. Gen. Virol. 83 (Pt 7): 1547–64. doi:10.1099/0022-1317-83-7-1547. PMID 12075073.
- ↑ Wang, X.W. Gibson, M.K. Vermeulen, W. Yeh, H. Forrester, K. Sturzbecher, H.W. Hoeijmakers, J.H. and Harris, C.C. (1995). "Abrogation of p53-induced Apoptosis by the Hepatitis B Virus X Gene". Cancer Res. 55 (24): 6012–6. PMID 8521383.
- ↑ Cyrelys Collazo; Osmani Chacón; Orlando Borrás (2006). "Programmed cell death in plants resembles apoptosis of animals" (PDF). Biotecnología Aplicada. 23: 1–10.
- ↑ Xiang J. et al., BAX-induced cell death may not require interleukin 1β-converting enzyme-likeproteases, 1996, cell biology
- ↑ Zhou, G. P. & Doctor, K. (2003). Subcellular location prediction of apoptosis proteins. PROTEINS: Structure, Function, and Genetics 50, 44-48.
- ↑ Chen Y. L.; Li Q. Z. (2007). "Prediction of apoptosis protein subcellular location using improved hybrid approach and pseudo amino acid composition". Journal of Theoretical Biology. 248 (2): 377–381. doi:10.1016/j.jtbi.2007.05.019. PMID 17572445.
- ↑ Ding, Y. S. & Zhang, T. L. (2008). Using Chou's pseudo amino acid composition to predict subcellular localization of apoptosis proteins: an approach with immune genetic algorithm-based ensemble classifier. Pattern Recognition Letters 29, 1887-1892.
- ↑ Jiang, X., Wei, R., Zhang, T. L. & Gu, Q. (2008). Using the concept of Chou's pseudo amino acid composition to predict apoptosis proteins subcellular location: an approach by approximate entropy. Protein & Peptide Letters 15, 392-396.
- ↑ Lin H, Wang H, Ding H, Chen YL, Li QZ (2009). "Prediction of Subcellular Localization of Apoptosis Protein Using Chou's Pseudo Amino Acid Composition". Acta Biotheoretica. 57: 321–330. doi:10.1007/s10441-008-9067-4.
- ↑ "Atromentin-Induced Apoptosis in Human Leukemia U937 Cells". Kim Jin Hee and Choong Hwan Lee, Journal of Microbiology and Biotechnology, 2009, vol. 19, no. 9, pages 946–950, INIST:21945937
Bibliography
- Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Morgan, David; Raff, Martin; Roberts, Keith; Walter, Peter (2015). Molecular Biology of the Cell (6th ed.). Garland Science. p. 2. ISBN 978-0815344322.
External links
- Apoptosis & cell surface
- Apoptosis & Caspase 3, The Proteolysis Map—animation
- Apoptosis & Caspase 8, The Proteolysis Map—animation
- Apoptosis & Caspase 7, The Proteolysis Map—animation
- Apoptosis MiniCOPE Dictionary—list of apoptosis terms and acronyms
- Apoptosis (Programmed Cell Death) – The Virtual Library of Biochemistry and Cell Biology
- Apoptosis Research Portal
- Apoptosis Info Apoptosis protocols, articles, news, and recent publications.
- Database of proteins involved in apoptosis
- Apoptosis Video
- Apoptosis Video (WEHI on YouTube )
- The Mechanisms of Apoptosis Kimball's Biology Pages. Simple explanation of the mechanisms of apoptosis triggered by internal signals (bcl-2), along the caspase-9, caspase-3 and caspase-7 pathway; and by external signals (FAS and TNF), along the caspase 8 pathway. Accessed 25 March 2007.
- WikiPathways – Apoptosis pathway
- Finding Cancer's Self-Destruct Button CR magazine (Spring 2007). Article on apoptosis and cancer.
- Xiaodong Wang's lecture: Introduction to Apoptosis
- Robert Horvitz's short clip: Discovering Programmed Cell Death
- The Bcl-2 Database
- DeathBase: a database of proteins involved in cell death, curated by experts
- European Cell Death Organization