Werner syndrome
| Werner syndrome (progeria) | |
|---|---|
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E34.8 (ILDS E34.820) |
| ICD-9-CM | 259.8 |
| OMIM | 277700 |
| DiseasesDB | 14096 |
| Patient UK | Werner syndrome |
| MeSH | C16.320.925 |
| GeneReviews | |
| Orphanet | 902 |
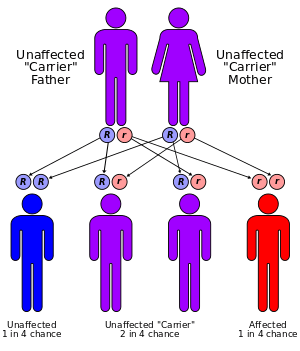
Werner syndrome (WS), also known as "adult progeria",[1] is a rare, autosomal recessive[2][3][4] progeroid syndrome (PS), which is characterized by the appearance of premature aging.[4]
Werner syndrome is named after the German scientist Otto Werner.[5] He identified the syndrome in four siblings observed with premature aging, which he explored as the subject of his dissertation of 1904.[6]
It has a global incidence rate of less than 1 in 100,000 live births[3] (although incidence in Japan and Sardinia is higher, affecting 1 in 20,000–40,000 and 1 in 50,000, respectively).[7][8] 1,300 cases had been reported as of 2006.[9] Affected individuals typically grow and develop normally until puberty; the mean age of diagnosis is twenty-four, often realized when the adolescent growth spurt is not observed.[10] The youngest person diagnosed was six years old.[11] The median and mean ages of death are 47–48 and 54 years, respectively.[12] The main cause of death is cardiovascular disease or cancer.[9][10]
Background and history
Otto Werner was the first to observe Werner syndrome in 1904 as a part of his dissertation research. As a German ophthalmologist, Werner described several progeria-like features and juvenile cataracts in many of his patients. He noticed these symptoms particularly in a family with four sequential children who all showed the characteristics of the syndrome at around the same age. He assumed the cause to be genetic, though most of his evidence was clinical. Between 1934 and 1941, two internists from New York, Oppenheimer and Kugel, coined the term "Werner Syndrome," igniting a wave of interest and research on the disease.[13] During that time, Agatson and Gartner suggested a possible link between Werner's syndrome and cancer. However, It was not until 1966 that there was a general consensus on the autosomal recessive mode of inheritance for the syndrome. By 1981, geneticists had located the WRN gene on chromosome 8, leading to its cloning in 1996. This cloning of the WRN was significant because it revealed the predicted WRN protein was made from a family of DNA helicases.[13] Prior to 1996, Werner syndrome was thought to be a model for accelerated aging. Since the discovery of the gene, it has become clear that the premature aging displayed in Werner syndrome is not the same, on a cellular level, as normal aging. The role of WRN in DNA repair and its exonuclease and helicase activities have been the subject of many studies in recent years.[14]
Since the initial discovery in 1904, several other cases of Werner syndrome have been recorded. Many of these cases have occurred in Japan, where a founder effect has caused a higher incidence rate than in other populations. The incidence rate of Werner syndrome in Japan is approximately 1 case per 100 thousand people (1:100,000), a large contrast with the rate of incidence for the rest of the world, which is between 1:1,000,000 and 1:10,000,000. A founder effect is also apparent in Sardinia, where there have been 18 recorded cases of Werner syndrome.[15]
Characteristics
Werner syndrome patients exhibit growth retardation, short stature, premature graying of hair, alopecia (hair loss), wrinkling, prematurely aged faces with beaked noses, skin atrophy (wasting away) with scleroderma-like lesions, lipodystrophy (loss of fat tissues), abnormal fat deposition leading to thin legs and arms, and severe ulcerations around the Achilles tendon and malleoli (around ankles). Other symptoms include change in voice (weak, hoarse, high-pitched), atrophy of gonads leading to reduced fertility, bilateral cataracts (clouding of lens), premature arteriosclerosis (thickening and loss of elasticity of arteries), calcinosis (calcium deposits in blood vessels), atherosclerosis (blockage of blood vessels), type 2 diabetes, osteoporosis (loss of bone mass), telangiectasia, and malignancies.[3][9] The prevalence of rare cancers, such as meningiomas, are increased in individuals with Werner syndrome.[16]
Diagnosis and clinical symptoms
The mutation in the WRN gene that causes Werner syndrome is autosomal and recessive, meaning that sufferers must inherit a copy of the gene from each parent. Patients display rapid premature aging beginning in young adulthood, usually in their early twenties.[17] Diagnosis is based on six cardinal symptoms: premature graying of the hair or hair loss, presence of bilateral cataracts, atrophied or tight skin, soft tissue calcification, sharp facial features, and an abnormal, high-pitched voice.[15] Patients are also generally short-statured due to absence of the adolescent growth spurt. Patients also display decreased fertility.[18] The most common symptom of the six is premature graying and loss of hair. This is also generally the earliest observed symptom, with hair loss occurring first on the scalp and the eyebrows.[18]
Werner syndrome patients often have skin that appears shiny and tight, and may also be thin or hardened.[17][18] This is due to atrophy of the subcutaneous tissue and dermal fibrosis.[18] Over time, facial features may be more apparent due to these skin conditions. Other associated skin conditions include ulcers,[18] which are very difficult to treat in Werner syndrome patients, and are caused in part by decreased potential of skin cells for replication.[13]
WS cataracts are distinctly different from those of normal aging. They are associated with problems in the lens posterior cortex and subcapsular regions. These cataracts are generally treatable with cataract surgery, which should restore normal vision.[18]
Symptoms become apparent in the late teens and early twenties and continue to progress. Most patients live to about fifty years of age. The most common causes of death for people are associated diseases and complications, especially atherosclerosis and cancer.[17]
Associated diseases
Werner syndrome patients are at increased risk for several other diseases, many associated with aging. Atherosclerosis, the thickening of artery walls due to cholesterol buildup, is one common complication.[15] While normal atherosclerosis generally involves the major arteries, smaller arterioles are more likely to be affected.[14] It is possible nervous system disorders are associated. Brain atrophy is present in 40% of patients.[13][15] Osteoporosis, the loss of bone mineral density common in post-menopausal women, is another common symptom. In contrast with the normal population, the rate of osteoporosis is especially high for male patients.[13] Diabetes mellitus is another common accompaniment.[15] Skin ulcers occur in about 75% of patients – and can be difficult to treat. If skin ulcers become badly infected or develop gangrene, they often require amputation. Unlike most other related diseases and complications, these ulcers are not associated with normal aging.[13]
Patients are also at an increased risk of cancer, especially malignant melanoma.[13] Soft-tissue sarcomas are the most common cancer types.[18] Other types of skin cancer, other epithelial cancers such as thyroid and liver cancers, MDS (myelodysplastic syndrome), and MFH (malignant fibrous histiocytoma) are also prevalent among.[13] Mutations in the WRN gene, especially single-nucleotide polymorphisms (SNPs), are associated with many of the cancers and other associated diseases. WRN SNPs correlate with cancers such as sarcomas and non-Hodgkin lymphomas, as well as diabetes and cardiovascular problems including atherosclerosis.[19]
Causes
Approximately 90% of individuals presenting Werner syndrome have any of a range of mutations in the gene, WRN, the only gene currently attributed to cause Werner syndrome.[11][12] WRN, which lies on chromosome 8 in humans,[20] encodes the WRNp protein, a 1432 amino acid protein with a central domain resembling members of the RecQ helicases. RecQ helicases are a special type of helicase that function at unique times during DNA repair of doubled stranded breaks, which are a form of DNA damage that results in a break of both strands of DNA. Thus, RecQ helicases are important for maintaining DNA stability, and loss of function of these helicases has important implications in the development of Werner syndrome.[21] In addition to the central domain, there are three exonuclease domains at the N-terminus and a Helicase and Ribonuclease D C-terminal (HRDC) domain at the C-terminus.[22]
When functioning normally, the WRN gene and associated protein are important for maintaining genome stability.[14] WRNp is active in unwinding DNA, a step necessary in DNA repair and DNA replication.[4][7] Specifically, the WRN protein has an important role in responding to replication malfunctions, particularly double-stranded breaks, and stalled replication machinery.[14] WRN may reactivate replication by preventing unwanted recombination processes from occurring or by promoting recombination, depending on the type of DNA damage. In addition, the WRN protein physically interacts with or binds to several other proteins that are involved in processing DNA.[23] For example, the WRN protein binds to RPA, which stimulates WRNp's helicase activity. WRNp also physically interacts with p53, a tumor suppressor gene that stops the formation of tumors and the progression of cancers,[24] which inhibits the exonuclease activity of the WRNp.[25] Since WRNp's function depends on DNA, it is only functional when localized to the nucleus.
Effects on cell structure & function
Mutations which cause Werner syndrome all occur at the regions of the gene which encode for protein, and not at non-coding regions.[26] There are 35 different known mutations of WRN, which correspond to stop codons, insertions, or deletions that result in a frameshift mutation.[25] These mutations can have a range of effects. They may decrease the stability of the transcribed messenger RNA (mRNA), which increases the rate at which they are degraded. With less mRNA, less is available to be translated into the WRNp protein. Mutations may also lead to the truncation (shortening) of the WRNp protein, leading to the loss of its nuclear localization signal sequence, thus it is no longer transported into the nucleus where it interacts with the DNA. This leads to a reduction in DNA repair.[26] Furthermore, mutated proteins are more likely to be degraded than normal WRNp.[7] Apart from causing defects in DNA repair, its aberrant association with p53 down-regulates the function of p53, leading to a reduction in p53-dependent apoptosis and increasing the survival of these dysfunctional cells.[27] Cells of affected individuals also have reduced lifespan in culture,[28] have more chromosome breaks and translocations[29] and have extensive deletions.[30]
Patients with Werner syndrome lose the RecQ helicase activity in the WRN protein because of the loss of its C-terminus region, but the mechanism by which this happens is unclear. The loss of the helicase activity can have far-reaching consequences in terms of cell stability and mutation. One instance of these consequences involves telomeres. It is thought that the WRN helicase activity is important not only for DNA repair and recombination, but also for maintaining telomere length and stability. Thus, WRN helicase is important for preventing catastrophic telomere loss during DNA replication.[31] In a normal cell, the telomeres (the ends of chromosomes) undergo repeated shortening during the cell cycle, which can prevent the cell from dividing and multiplying. This event can be counteracted by telomerase, an enzyme that extends the ends of the chromosomes by copying the telomeres and synthesizing an identical, but new end that can be added to the existing chromosome.[32] However, patients with Werner syndrome often exhibit accelerated telomere shortening, indicating that there may be a connection between the loss of the WRN helicase activity and telomere and cell instability. While evidence shows that telomere dysfunction is consistent with the premature aging in WS, it has yet to be determined if it is the actual cause of the genomic instability observed in cells and the high rate of cancer in WS patients.[31]
Without the WRN protein, the interwoven pathways of DNA repair and telomere maintenance fail to suppress cancer and the aging symptoms seen in patients with WS. Events such as rapid telomere shortening cause Werner syndrome cells to exhibit low responses to overall cellular stress. In addition to telomere dysfunction, over-expression of oncogenes and oxidation can induce this type of response. High stress causes a synergistic effect, where WS cells become even more sensitive to agents that increase cell stress and agents that damage DNA. As a result, WS cells show a drastic reduction in replicative lifespan and enter into a stage of aging prematurely. The accumulation of these damaged cells due to telomere shortening over many years may be indicative of why Werner syndrome symptoms only appear after an individual is about twenty years old.[33]
Treatment
A cure for Werner syndrome has not yet been discovered. It is often treated by managing the associated diseases and relieving symptoms to improve quality of life. The skin ulcers that accompany WS can be treated in several ways, depending on the severity. Topical treatments can be used for minor ulcers, but are not effective in preventing new ulcers from occurring. In the most severe cases, surgery may be required to implant a skin graft or amputate a limb if necessary. Diseases commonly associated with Werner Syndrome such as diabetes and cancer are treated in generally the same ways as they would be for a non-Werner Syndrome individual. A change in diet & exercise can help prevent and control arteriosclerosis, and regular cancer screenings can allow for early detection of cancer.[34]
There is recent evidence that suggests that the cytokine-suppressive anti-inflammatory drug, SB203580, may be a possible therapeutic option for patients with Werner's Syndrome. This drug targets the p38 signaling pathway, which may become activated as a result of genomic instability and stalled replication forks that are characteristic mutations in WS. This activation of p38 may play a role in the onset of premature cell aging, skin aging, cataracts, and graying of the hair. The p38 pathway has also been implicated in the anti-inflammatory response that causes atherosclerosis, diabetes, and osteoporosis, all of which are associated with Werner's Syndrome. This drug has shown to revert the aged characteristics of young WS cells to those seen in normal, young cells and improve the lifespan of WS cells in vitro. SB203580 is still in the clinical trial stages, and the same results have not yet been seen in vivo.[35]
In 2010, vitamin C supplementation was found to reverse the premature aging and several tissue dysfunctions in a genetically modified mouse model of the disease. Vitamin C supplementation also appeared to normalize several age-related molecular markers such as the increased levels of the transcription factor NF-κB. In addition, it decreases activity of genes activated in human Werner syndrome and increases gene activity involved in tissue repair. Supplementation of vitamin C is suspected to be beneficial in the treatment of human Werner syndrome, although there was no evidence of anti-aging activity in nonmutant mice.[36] In general, treatments are available for only the symptoms or complications and not for the disease itself.[37]
Popular culture
On the episode "Stargazer in a Puddle" from the television series Bones, the victim has Werner syndrome.
Werner syndrome is featured in the film Jack, starring Robin Williams, in which his character ages four times faster than normal.
In an early cut scene from the game Metal Gear Solid 4, Otacon cites "classic Werner syndrome" as the most likely cause of Solid Snake's premature aging, though he goes on to say that testing had been inconclusive. It is later said that Solid Snake's body was designed to break down quickly.
In season 3 episode 9, "The Ballad of Kevin and Tess", of TV series The 4400, Kevin is said to have Werner syndrome to hide his real condition from the public.
In The Invisible Man season 1 episode 6, "Impetus", the new character Gloria has an experimentally altered type of Werner syndrome that causes it to become contagious.
The central character in Gail Tsukiyama's novel DREAMING WATER (2002) has Werner's syndrome.
In season 1 episode 8 Cold Comfort from TV series Dark Angel, a character has a "form of progeria, similar to Werner syndrome", due to genetic manipulation. With an appropriate treatment, her condition seems to be stabilized.
See also
References
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ↑ Ozgenc A, Loeb LA (Sep 2005). "Current advances in unraveling the function of the Werner syndrome protein". Mutat. Res. 577 (1-2): 237–51. doi:10.1016/j.mrfmmm.2005.03.020. PMID 15946710.
- 1 2 3 Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J (2003). "Aging and genome maintenance: lessons from the mouse?". Science. 299 (5611): 1355–9. doi:10.1126/science.1079161. PMID 12610296.
- 1 2 3 Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM, Oshima J, Loeb LA (1997). "The Werner syndrome protein is a DNA helicase". Nat. Genet. 17 (1): 100–3. doi:10.1038/ng0997-100. PMID 9288107.
- ↑ synd/892 at Who Named It?
- ↑ "On cataract in conjunction with scleroderma. Otto Werner, doctoral dissertation, 1904, Royal Ophthalmology Clinic, Royal Christian Albrecht University of Kiel". Adv. Exp. Med. Biol. 190: 1–14. 1985. doi:10.1007/978-1-4684-7853-2_1. PMID 3909762.
- 1 2 3 "Werner syndrome". Genetics Home Reference. Retrieved 18 March 2013.
- ↑ Masala MV, Scapaticci S, Olivieri C, Pirodda C, Montesu MA, Cuccuru MA, Pruneddu S, Danesino C, Cerimele D (2007). "Epidemiology and clinical aspects of Werner's syndrome in North Sardinia: description of a cluster". Eur J Dermatol. 17 (3): 213–6. doi:10.1684/ejd.2007.0155 (inactive 2015-01-23). PMID 17478382.
- 1 2 Epstein CJ, Martin GM, Schultz AL, Motulsky AG (1966). "Werner's syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process". Medicine (Baltimore). 45 (3): 177–221. doi:10.1097/00005792-196605000-00001. PMID 5327241.
- 1 2 Oshima J, Martin GM, Hisama FM (February 2012) [1993–]. "Werner Syndrome". In Pagon RA, Bird TD, Dolan CR, et al. GeneReviews™ [Internet]. Seattle WA: University of Washington, Seattle. PMID 20301687. NBK1514.
- 1 2 Oshima J, Martin GM, Hisama FM. Werner Syndrome. 2002 Dec 2 [Updated 2012 Dec 13]. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Available from:http://www.ncbi.nlm.nih.gov/books/NBK1514/
- 1 2 3 4 5 6 7 8 Goto, Makoto (2004). Clinical Aspects of Werner's Syndrome: Its Natural History and the Genetics of the Disease. Eurekah.com and Kluwer Academic / Plenum Publishing. p. 1.
- 1 2 3 4 Chen L, Huang S, Lee L, Davalos A, Schiestl RH, Campisi J, Oshima J (2003). "WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair". Aging Cell. 2 (4): 191–9. doi:10.1046/j.1474-9728.2003.00052.x. PMID 12934712.
- 1 2 3 4 5 Coppedè, Fabio (August 2013). "The epidemiology of premature aging and associated comorbidities". Clinical Interventions in Aging. p. 1023. doi:10.2147/CIA.S37213.
- ↑ Goto M, Miller RW, Ishikawa Y, Sugano H (1996). "Excess of rare cancers in Werner syndrome (adult progeria)". Cancer Epidemiol. Biomarkers Prev. 5 (4): 239–46. PMID 8722214.
- 1 2 3 "Genetics Home Reference". US National Library of Medicine.
- 1 2 3 4 5 6 7 Monnat RJ (October 2010). "Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology". Semin. Cancer Biol. 20 (5): 329–39. doi:10.1016/j.semcancer.2010.10.002. PMC 3040982
 . PMID 20934517.
. PMID 20934517. - ↑ Rossi ML, Ghosh AK, Bohr VA (March 2010). "Roles of Werner syndrome protein in protection of genome integrity". DNA Repair (Amst.). 9 (3): 331–44. doi:10.1016/j.dnarep.2009.12.011. PMC 2827637. PMID 20075015.
- ↑ Goto M, Rubenstein M, Weber J, Woods K, Drayna D (Feb 1992). "Genetic linkage of Werner's syndrome to five markers on chromosome 8". Nature. 355 (6362): 735–8. Bibcode:1992Natur.355..735G. doi:10.1038/355735a0. PMID 1741060.
- ↑ Bernstein KA, Gangloff S, Rothstein R (2010). "The RecQ DNA helicases in DNA repair". Annu. Rev. Genet. 44: 393–417. doi:10.1146/annurev-genet-102209-163602. PMC 4038414. PMID 21047263.
- ↑ Chen L, Oshima J (2002). "Werner Syndrome". J. Biomed. Biotechnol. 2 (2): 46–54. doi:10.1155/S1110724302201011. PMC 153784. PMID 12488583.
- ↑ Pichierri P, Ammazzalorso F, Bignami M, Franchitto A (March 2011). "The Werner syndrome protein: linking the replication checkpoint response to genome stability". Aging (Albany NY). 3 (3): 311–8. PMC 3091524. PMID 21389352.
- ↑ Harris CC (1996). "Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies". J. Natl. Cancer Inst. 88 (20): 1442–55. doi:10.1093/jnci/88.20.1442. PMID 8841019.
- 1 2 Opresko PL, Cheng WH, von Kobbe C, Harrigan JA, Bohr VA (2003). "Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process". Carcinogenesis. 24 (5): 791–802. doi:10.1093/carcin/bgg034. PMID 12771022.
- 1 2 Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, Poot M, Rubin CD, Chen DF, Yang CC, Juch H, Dorn T, Spiegel R, Oral EA, Abid M, Battisti C, Lucci-Cordisco E, Neri G, Steed EH, Kidd A, Isley W, Showalter D, Vittone JL, Konstantinow A, Ring J, Meyer P, Wenger SL, von Herbay A, Wollina U, Schuelke M, Huizenga CR, Leistritz DF, Martin GM, Mian IS, Oshima J (2006). "The spectrum of WRN mutations in Werner syndrome patients". Hum. Mutat. 27 (6): 558–67. doi:10.1002/humu.20337. PMC 1868417. PMID 16673358.
- ↑ Spillare EA, Robles AI, Wang XW, Shen JC, Yu CE, Schellenberg GD, Harris CC (1999). "p53-mediated apoptosis is attenuated in Werner syndrome cells". Genes Dev. 13 (11): 1355–60. doi:10.1101/gad.13.11.1355. PMC 316776. PMID 10364153.
- ↑ Martin GM, Sprague CA, Epstein CJ (1970). "Replicative life-span of cultivated human cells. Effects of donor's age, tissue, and genotype". Lab. Invest. 23 (1): 86–92. PMID 5431223.
- ↑ Salk D, Au K, Hoehn H, Martin GM (1981). "Cytogenetics of Werner's syndrome cultured skin fibroblasts: variegated translocation mosaicism". Cytogenet. Cell Genet. 30 (2): 92–107. doi:10.1159/000131596. PMID 7273860.
- ↑ Fukuchi K, Martin GM, Monnat RJ (1989). "Mutator phenotype of Werner syndrome is characterized by extensive deletions". Proc. Natl. Acad. Sci. U.S.A. 86 (15): 5893–7. Bibcode:1989PNAS...86.5893F. doi:10.1073/pnas.86.15.5893. PMC 297737. PMID 2762303.
- 1 2 Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J (Feb 13, 2007). "Telomere dysfunction as a cause of genomic instability in Werner syndrome". Proc. Natl. Acad. Sci. U.S.A. 104 (7): 2205–10. Bibcode:2007PNAS..104.2205C. doi:10.1073/pnas.0609410104. JSTOR 25426449. PMID 17284601.
- ↑ Chan SR, Blackburn EH (2004). "Telomeres and telomerase". Philos. Trans. R. Soc. Lond., B, Biol. Sci. 359 (1441): 109–21. doi:10.1098/rstb.2003.1370. PMID 15065663.
- ↑ Multani AS, Chang S (December 2006). "WRN at telomeres: implications for aging and cancer". J. Cell. Sci. 120 (Pt 5): 713–21. doi:10.1242/jcs.03397. PMID 17314245. Check date values in:
|year= / |date= mismatch(help) - ↑ "Werner's Syndrome: Treatments". Progeria Information Database. Retrieved April 12, 2014.
- ↑ Davis T, Kipling D (2006). "Werner Syndrome as an example of inflamm-aging: possible therapeutic opportunities for a progeroid syndrome?". Rejuvenation Res. 9 (3): 402–7. doi:10.1089/rej.2006.9.402. PMID 16859481.
- ↑ Massip L, Garand C, Paquet ER, Cogger VC, O'Reilly JN, Tworek L, Hatherell A, Taylor CG, Thorin E, Zahradka P, Le Couteur DG, Lebel M (2009-09-09). "Vitamin C restores healthy aging in a mouse model for Werner syndrome". FASEB J. 24 (1): 158–72. doi:10.1096/fj.09-137133. PMID 19741171. Check date values in:
|year= / |date= mismatch(help) - ↑ Coppedè F (August 5, 2013). "The epidemiology of premature aging and associated comorbidities". Clin Interv Aging. 8: 1023–32. doi:10.2147/CIA.S37213. PMC 3760297. PMID 24019745.
External links
This article incorporates public domain text from The U.S. National Library of Medicine
- Werner Syndrome from GeneReviews™, contains extensive information on the disorder
- Werner syndrome at libero.it
- DNA Repair at rcn.com
- Segmental Progeria at benbest.com
- WRN at the GenAge database.
- werner at NIH/UW GeneTests
- Werner Syndrome at 00421 at CHORUS
