Xeroderma pigmentosum
| Xeroderma pigmentosum | |
|---|---|
 | |
| An eight-year-old girl from Guatemala with xeroderma pigmentosum[1] | |
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q82.1 |
| ICD-9-CM | 757.33 |
| OMIM | 278700 |
| DiseasesDB | 14198 |
| MedlinePlus | 001467 |
| eMedicine | derm/462 neuro/399 |
| Patient UK | Xeroderma pigmentosum |
| MeSH | D014983 |
| GeneReviews | |
| Orphanet | 910 |

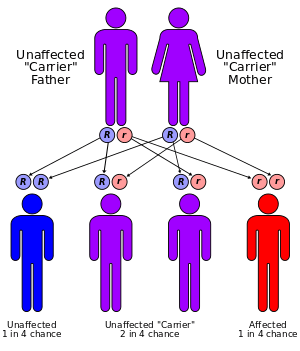
Xeroderma pigmentosum (XP) is a rare autosomal recessive genetic disorder of DNA repair in which the ability to repair damage caused by ultraviolet (UV) light is deficient.[2]:574 In extreme cases, all exposure to sunlight must be forbidden, no matter how small; as such, individuals with the disease are often colloquially referred to as "Moon child".[3] Multiple basal cell carcinomas (basaliomas) and other skin malignancies frequently occur at a young age in those with XP; metastatic malignant melanoma and squamous cell carcinoma[4] are the two most common causes of death in XP victims. This disease is present in both genders and in all races, with an incidence of 1:250,000 in the United States.[5] XP is roughly six times more common in Japanese people[4] than in other groups.
Normally, damage to DNA in epidermal cells occurs during exposure to UV light. The absorption of the high-energy light leads to the formation of pyrimidine dimers, namely cyclobutane-pyrimidine dimers and pyrimidine-6-4-pyrimidone photoproducts. In a healthy, normal human being, the damage is first excised by endonucleases. DNA polymerase then repairs the missing sequence, and ligase "seals" the transaction. This process is known as nucleotide excision repair.
Genetics
One of the most frequent defects in xeroderma pigmentosum is an autosomal recessive genetic defect in which nucleotide excision repair (NER) enzymes are mutated, leading to a reduction in or elimination of NER.[6] If left unchecked, damage caused by ultraviolet light can cause mutations in individual cell's DNA. The causes of the neurological abnormalities are poorly understood and are not connected with exposure to ultraviolet light. The most current theories suggest that oxidative DNA damage is generated during normal metabolism in the central nervous system, and that some types of this damage must be repaired by NER[7]
Since DNA repair is under genetic control, it can easily undergo mutations. Many genetic disorders such as xeroderma pigmentosum (XP; MIM 278700) are caused by mutations in genes that repair DNA.[7] If the gene was not repaired correctly it could cause xeroderma pigmentosum in individuals. The autosomal recessive disorder xeroderma pigmentosum or XP has a frequency of 1 in every 250,000 individuals of all races and ethnic groups.[8] Those affected with the autosomal recessive disorder XP are extremely sensitive to UV light produced by the sun and even with a short exposure to it causes dry, flaking skin and pigmented spots that can develop into skin cancer. Individuals with XP are about 1,000 times more likely to develop skin cancer than individuals without the disorder.
The molecular defects in XP cells result in a greatly elevated induction of mutations in sun-exposed skin of affected individuals. This increased mutation frequency probably accounts for the pigmentation changes and the skin cancers. Examination of mutations in the p53 gene in tumors from XP patients reveal p53 mutations characteristic of UV exposure in the majority of tumors[8] As with all genetic disorders, genetic counseling and psychological support is appropriate for the families, to discuss probability of occurrence in future pregnancies, feelings of isolation and concern about career prospects. Although there is no cure for xeroderma pigmentosum, the effects can be minimized by getting protection from the sunlight and if possible early removal of precancerous lesions. The most common fate for individuals with XP is early death from cancer due to the fact that they need to take outstanding measures to protect themselves from the dangers of the UV light. But if there is an absence of neurological problems and the individual is always protected or away from sunlight, the prognosis is good.
Types
There are seven complementation groups, plus one variant form:
| Type | Diseases Database | OMIM | Gene | Locus | Also known as / description |
| Type A, I, XPA | 29877 | 278700 | XPA | 9q22.3 | Xeroderma pigmentosum group A - the classical form of XP |
| Type B, II, XPB | 29878 | 133510 | XPB | 2q21 | Xeroderma pigmentosum group B |
| Type C, III, XPC | 29879 | 278720 | XPC | 3p25 | Xeroderma pigmentosum group C |
| Type D, IV, XPD | 29880 | 278730 278800 | XPD ERCC6 | 19q13.2-q13.3, 10q11 | Xeroderma pigmentosum group D or De Sanctis-Cacchione syndrome (can be considered a subtype of XPD) |
| Type E, V, XPE | 29881 | 278740 | DDB2 | 11p12-p11 | Xeroderma pigmentosum group E |
| Type F, VI, XPF | 29882 | 278760 | ERCC4 | 16p13.3-p13.13 | Xeroderma pigmentosum group F |
| Type G, VII, XPG | 29883 | 278780 133530 | RAD2 ERCC5 | 13q33 | Xeroderma pigmentosum group G and COFS syndrome type 3 |
| Type V, XPV | 278750 | POLH | 6p21.1-p12 | Xeroderma pigmentosum variant - these patients suffer from mutation in a gene that codes for a specialized DNA polymerase called polymerase-η (eta). Polymerase-η can replicate over the damage and is needed when cells enter S-phase in the presence of a DNA-replication. |
Symptoms
Symptoms include:
- Severe sunburn when exposed to only small amounts of sunlight. These often occur during a child's first exposure to sunlight.
- Development of many freckles at an early age
- Rough-surfaced growths (solar keratoses), and skin cancers
- Eyes that are painfully sensitive to the sun and may easily become irritated, bloodshot and clouded
- Blistering or freckling on minimum sun exposure
- Spider Veins
- Limited growth of hair on chest and legs
- Scaly skin
- Dry skin
- Irregular dark spots on the skin
- Corneal ulcerations
Treatment
The most obvious, and often important part of treatment, is avoiding exposure to sunlight. This includes wearing protective clothing and using sunscreen (physical and chemical).[9] Keratoses can also be treated using cryotherapy or fluorouracil.[1]
Prognosis
Fewer than 40% of individuals with the disease survive beyond the age of 20. Some XP victims with less severe cases do manage to live well into their 40s.
In popular culture
These fictional characters have XP:
- Christopher Snow in Dean Koontz's Moonlight Bay Trilogy
- 1994 CBS-TV movie Children of the Dark is based on XP.
- Luke in the 2002 novel Going Out by Scarlett Thomas
- In the Channel 4 series Ultraviolet, one of the humans is mistaken for a vampire because he avoids sunlight, when in fact he has XP.
- In the book The Lucifer Code (originally published in 1997 as "Lucifer") the character Bradley Soames suffers from XP, and wears an all-encompassing "suit" complete with mask, hood and heavily tinted lenses in order to venture outdoors.
- In the independent film Dark Side of the Sun (1988) with Brad Pitt as the main character suffering from XP.
- In the 2001 film The Others, the two children, Anne and Nicholas, suffer from XP.
- In the 2003 novel Second Glance by Jodi Picoult, Ethan Wakeman, the 9-year-old nephew of Ross Wakeman (the main protagonist) has XP.
- The 2003 Angela Johnson novel, A Cool Moonlight, centers on a girl who has XP and can never be in the sun. The family has gone to drastic measures to help make her life easier, and to make her feel like a normal 8-year-old.
- The 2006 Japanese drama "Taiyou no uta" (A Song to the Sun) centers around a girl with XP who dreams of being a singer.
- The 2006 German film Mondscheinkinder (Children of the Moonlight) features 12-year-old Lisa who creates a fantasy world for her 6-year-old brother Paul, who has XP and cannot leave the house. Their special relationship is threatened when Lisa gets her first boyfriend, facing her with hard choices.[10]
- The Spanish film "Eskalofrío" or "Shiver" released in 2008 featured a main character named Santi who is ostracized as he suffers from the condition.
- The 2011 film La permission de minuit by French director Delphine Gleize centers on a teenage boy with XP.
- The 2012 documentary "Sun Kissed" explores the XP problem on the Navajo Indian Reservation.
- The 2013 young adult novel What We Saw at Night by Jacquelyn Mitchard tells the story of three teenagers who are suffering from this disease, go out only when dark and witness something strange one night.
- The 2013 middle grade novel "Doom & Gloom" by M. J. Shaughnessy tells the story of a twelve-year-old boy who triumphs over his disease by donning a protective solar suit and becoming a superhero.
- The 2014 adult fiction book The Deepest Secret by Carla Buckley tells the story of a mother with a son who has XP and the lengths she will go to in order to protect him from a crisis that threatens to tear the community apart.[11]
- In the 2016 horror film Lights Out (2016 film), the villain, Diana had this condition. She was killed when the doctors at the mental institution she and Sophie stayed at tried treating the condition, but it went awry.
See also
- Biogerontology
- Cockayne syndrome
- List of cutaneous conditions
- List of cutaneous conditions associated with increased risk of nonmelanoma skin cancer
- Photophobia
- Senescence
References
- 1 2 Halpern, J.; Hopping, B.; Brostoff, J. (2008). "Photosensitivity, corneal scarring and developmental delay: Xeroderma Pigmentosum in a tropical country". Cases journal. 1 (1): 254. doi:10.1186/1757-1626-1-254. PMC 2577106
 . PMID 18937855.
. PMID 18937855. - ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ↑ Medical Biochemistry at a Glance. John Wiley & Sons. 28 November 2011. ISBN 1118292405. Retrieved 17 June 2011.
Xeroderma pigmentosa is a rare, autosomal recessive disease caused by a defective UV-specific endonuclease. Patients with mutations are unable to repair DNA damage caused by sunlight and have been described as "children of the night."
- 1 2 Li, Lei (January 8, 2007). "Chapter 3 Nucleotide Excision Repair". DNA REPAIR, GENETIC INSTABILITY, AND CANCER. World Scientific Publishing. pp. 75–76. ISBN 981-270-014-5.
- ↑ Lehmann AR, McGibbon D, Stefanini M (2011). "Xeroderma pigmentosum". Orphanet J Rare Dis. 6: 70. doi:10.1186/1750-1172-6-70. PMC 3221642. PMID 22044607.
- ↑ E. C. Friedberg; G. C. Walker; W. Siede; R. D. Wood; R. A. Schultz; T. Ellenberger (2006). DNA repair and mutagenesis. Washington: ASM Press. p. 1118. ISBN 978-1-55581-319-2.
- 1 2 Brooks PJ DNA Repair (Amst). 2014 March 2; 7(7):1168-79.
- 1 2 Daya-Grosjean L, Sarasin A Mutat Res. 2014 March 2; 571(1-2):43-56
- ↑ Nussbaum, Robert; McInnes, Roderick; Willard, Huntington. Genetics in Medicine. Elsevier. ISBN 978-14377-0696-3.
- ↑ See the website of the movie, in German: http://www.mondscheinkinder-der-film.de/
- ↑ https://www.goodreads.com/book/show/18248415-the-deepest-secret?ac=1
External links
| Wikimedia Commons has media related to Xeroderma pigmentosum. |
- Information
- GeneReviews/NCBI/NIH/UW entry on xeroderma pigmentosum
- An article about this disorder from DigiLander.iol.it (in English and Italian)
- Cancer.Net Xeroderma Pigmentosum
- DermNet systemic/xeroderma-pigmentosum
- Charities
- Short films
- Sloan Science and Film / Short Films / XP by David Barba 10 minutes
- Web Site of a Short Film about an xeroderma pigmentosum (XP) Patient. Film is directed by Kimberly Williams-Paisley