Pyroptosis
Pyroptosis is a highly inflammatory form of programmed cell death that occurs most frequently upon infection with intracellular pathogens and is likely to form part of the antimicrobial response. In this process, immune cells recognize foreign danger signals within themselves, release pro-inflammatory cytokines, swell, burst and die. The released cytokines attract other immune cells to fight the infection and contribute to inflammation in the tissue. Pyroptosis promotes the rapid clearance of various bacterial and viral infections by removing intracellular replication niches and enhancing the host’s defensive responses. However, in pathogenic chronic diseases, the inflammatory response does not eradicate the primary stimulus, as would normally occur in most cases of infection or injury, and thus a chronic form of inflammation ensues that ultimately contributes to tissue damage. Some examples of pyroptosis include salmonella-infected macrophages[1] and abortively HIV-infected T helper cells.[2][3]
The initiation of pyroptosis in infected macrophages is caused by the recognition of flagellin components of Salmonella and Shigella species (and similar pathogen-associated molecular patterns (PAMPs) in other microbial pathogens) by NOD-like receptors (NLRs). These receptors function like plasma membrane Toll-like receptors (TLRs), but recognise antigens located within the cell rather than outside of it.
In contrast to apoptosis, pyroptosis requires the function of the enzyme caspase-1.[4] Caspase-1 is activated during pyroptosis by a large supramolecular complex termed the pyroptosome (also known as an inflammasome).[5] Only one large pyroptosome is formed in each macrophage, within minutes after infection. Biochemical and Mass Spectroscopic analysis revealed that this pyroptosome is largely composed of dimers of the adaptor protein ASC (apoptosis-associated speck protein containing a CARD or Caspase activation and recruitment domain).
Unlike apoptosis, cell death by pyroptosis results in plasma-membrane rupture and the release of damage-associated molecular pattern (DAMP) molecules such as ATP, DNA and ASC oligomers (specks) into the extracellular milieu, including cytokines that recruit more immune cells and further perpetuate the inflammatory cascade in the tissue.[6][7] These processes are in marked contrast to the packaging of cellular contents and non-inflammatory phagocytic uptake of membrane-bound apoptotic bodies that characterizes apoptosis.
Discovery of pyroptosis
This type of inherently proinflammatory programmed cell death was named ‘pyroptosis’ in 2001 by Dr. Brad T.Cookson, an associate professor of microbiology and laboratory medicine of University of Washington.[8] "Pyro" and "ptosis" are two Greek words, the first word refers to fire and the latter means falling. The apparent meaning of the combined word "pyroptosis" is therefore, "the falling of fire", which here refers to the process of pro-inflammatory chemical signals bursting out of a host cell. Pyroptosis has a distinct morphology and mechanism compared to other forms of cell death. However, this form of cell death is akin to necrosis.[9] It was suggested that microbial-infection was the main evolutionary pressure for this pathway.[10]
Molecular Mechanism and Morphology
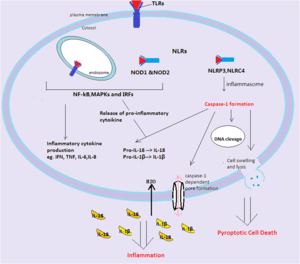
Infection can launch a ‘self-destruct’ and warning system in the host cell. Two types of receptors that belong to different families of pattern recognition receptors (PRRs) are present in the pyroptosis to sense intracellular and extracellular ‘danger’ signals. These are Nod-like receptors (NLRs) and Toll-like receptors (TLRs).[11] The ‘danger’ signals can be given off by invasive pathogens, or by an injury to a tissue, which can all be recognised by the host cells’ receptors.[12] That recognition will determine the fate of the host cell by a distinct mechanism, i.e. it will induce either the production of inflammatory chemical messengers termed ‘cytokines’ or programmed cell death. Commonly found cytokines are tumour necrosis factor (TNF), IL-6, IL-8, type I interferons (IFNs) and Interferon regulatory factor (IRFs). The inflammatory response is cell-death independent.[13]
In terms of cell death, although the activation route of caspase-1 is varied, the downstream signalling pathway will converge to result in the pyroptotic cell death. Cell lysis occurs upon the formation of pores, of an estimated diameter of 1.1-2.4 nm, in the cell membrane, which disrupts the cellular ionic gradient. The resulting increase in osmotic pressure causes an influx of water followed by cell swelling and bursting. At the same time, the cytosolic contents release via the channels of the pores.[14] The process is much like punctures in a water balloon. Subsequently, the inactive pro-inflammatory cytokines are further cleaved by caspase-1 and become activated.[15] Moreover, DNA cleavage with retained integrity and nuclear condensation has also been found to be associated with the process.
| Characteristics | Apoptosis | Pyroptosis | Necrosis | |
|---|---|---|---|---|
| Morphology | Cell lysis | NO | YES | YES |
| Cell swelling | NO | YES | YES | |
| Pore formation | NO | YES | YES | |
| Membrane blebbing | YES | NO | NO | |
| DNA fragmentation | YES | YES | YES | |
| Mechanism | Caspase-1 | NO | YES | NO |
| Caspase-3 | YES | NO | NO | |
| Cytochome-c release | YES | NO | NO | |
| Outcome | Inflammation | NO(anti) | YES | YES |
| Programmed cell death | YES | YES | NO |
Factors that are involved in the pathway
TLRs
Toll like receptors(TLRs) recognise Pathogen-Associated Molecular Patterns (PAMPs) that are located either in cell surface or within endosomes. The resulting recognition will initiate the signalling pathway, including the activation of transcription factors NF-κB and MAPKs. This in turn will be responsible for the production of inflammatory cytokines such as IFN α/β, TNF and IL-12. In addition, pro-IL-1β and pro-IL-18 will be released to be processed by cysteine-mediated caspase-1.[16]
NLRs
NLRs consist of more than 20 subsets,[17] including NOD1 and NOD2, NLRP3(also known as NALP3), NLRC4.[18] All recognise bacterial, viral and toxic foreign products that are introduced into the host cell cytosol. Upon recognition, NOD1 and NOD2 function similarly to the TLRs, producing and processing inflammatory cytokines.[19] Some of these subsets such as NLRP3 could also activate caspase-1 dependent cell death, accompanied by pore-forming and further stimulated by cellular potassium efflux. NLRC4 can specifically recognise flagellin and then trigger caspase-1 dependent pyroptosis.[11] NOD’s recognise molecular pattern danger signals and build up the inflammasome.
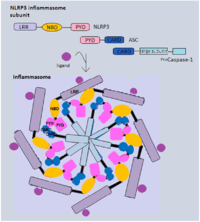
Inflammasome
The formation of the multi-protein complex inflammasome is achieved through the binding of intracellular bacterial, viral or host danger signals to the NLRs receptor, whose assembly leads to the activation of caspase-1[20] that is required in the processing and secretion of the pro-inflammatory cytokines. The best characterized inflammasome complex, NLRP3, has 3 distinct domains: several leucine-rich repeat (LRR) domains, a central nucleotide-binding and oligomerization domain (NBD) and an N-terminal pyrin domain(PYD).[17][19] The interaction between NLRP3 and caspase-1 is via the adaptor protein ASC. ASC contains a caspase activation and recruitment domain (CARD) that binds and facilitates activation of pro-caspase-1 through CARD-CARD interactions.[17] In some cases, NLRC4 can directly recruit caspase-1 as it has a CARD domain.
Caspase-1 Activation
The crucial enzyme required in the stimulation of the downstream pathway is caspase-1, which is located inside the cells. Caspase-1 was known as an interleukin-1β converting enzyme, as it was first discovered in association with the cleavage of pro-IL-1β.[14][21] The pro-caspase-1 with a 10-KDa CARD domain[22] will be recruited by various inflammasome. Similar to other caspases, caspase-1 starts off as an inactive precursor called zymogen. The caspase-1 enzymes become activated when they oligomerise and form tetramers. This process is spontaneous due to the fact that everything in the inflammasome is in close proximity with each other.[22] The cysteine-cleaved enzyme will not only cause cell death but is also responsible for the cleavage of the pro-inflammatory cytokines IL-1β and IL-18. The cytokines, once processed, will be in their biologically active form ready to be released from the host cells. The development of efficient adaptive immune responses depends on the recruitment and activation of the immune cells by inflammatory cytokines.
Clinical relevance
Pyroptosis acts as a defence mechanism against infection by inducing pathological inflammation. The formation of inflammasome and the activity of caspase-1 determine the balance between pathogen resolution and disease pathology.
In a healthy cell, caspase-1 activation helps to fight infection caused by Salmonella, Shigella via introducing cell death to restrict pathogen growth.[14] When the ‘danger’ signal is sensed, the quiescent cells will be activated to undergo pyroptosis and produce inflammatory cytokines IL-1β and IL-18. IL-18 will stimulate IFNγ production and initiates the development of TH1 responses. (TH1 responses tend to release cytokines that direct an immediate removal of the pathogen).[23] The cell activation results in an increase in cytokine levels, which will augment the consequences of inflammation and this, in turn, contributes to the development of the adaptive response as infection progresses. The ultimate resolution will clear pathogens.
In contrast, persistent inflammation will produce excessive immune cells which will be detrimental. If the amplification cycles persist, metabolic disorder, autoinflammatory diseases and liver injury associated with chronic inflammation will take place.[24]
Metabolic disorder
The level of expression of NLRP3 inflammasome and caspase-1 has direct relation with the severity of several metabolic syndromes, such as obesity and type II diabetic mellitus (T2DM). This is because the subsequent production level of IL-1β and IL-18, cytokines that impairs the secretion of insulin, is affected by the activity of caspase-1. Glucose uptake level is then diminished, and the condition is known as insulin resistance.[25] The condition is further accelerated by the IL-1β induced destruction of pancreatic β cell.[26]
Cryopyrinopathies
A mutation in the gene coding of inflammasome leads to a group of autoinflammatory disease called cryopyrinopathies. This group includes Muckle-Wells syndrome, cold autoinflammatory syndrome, and chronic infantile neurologic cutaneous and articular syndrome, all showing symptoms of sudden fevers and localized inflammation.[27] The mutated gene in this case is the NLRP3, impeding the activation of inflammasome, resulting in an excessive production IL-1β. This effect is known as "gain-of-function".[28]
HIV and AIDS
Recent studies demonstrated that caspase-1-mediated pyroptosis drives CD4 T-cell depletion and inflammation by HIV,[29][30] two signature events that propel HIV disease progression to AIDS. Although pyroptosis contributes to the host’s ability to rapidly limit and clear infection by removing intracellular replication niches and enhancing defensive responses through the release of proinflammatory cytokines and endogenous danger signals, in pathogenic inflammation, such as that elicited by HIV-1, this beneficial response does not eradicate the primary stimulus. In fact, it appears to create a pathogenic vicious cycle in which dying CD4 T cells release inflammatory signals that attract more cells into the infected lymphoid tissues to die and to produce chronic inflammation and tissue injury. It may be possible to break this pathogenic cycle with safe and effective caspase-1 inhibitors. These agents could form a new and exciting ‘anti-AIDS’ therapy for HIV-infected subjects in which the treatment targets the host instead of the virus. Of note, Caspase-1 deficient mice develop normally,[31][32] arguing that inhibition of this protein would produce beneficial rather than harmful therapeutic effects in HIV patients.
References
- ↑ Fink SL and Cookson BT (2006) "Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages". Cell Microbiol..
- ↑ Doitsh, Gilad; Galloway, Nicole L. K.; Geng, Xin; Yang, Zhiyuan; Monroe, Kathryn M.; Zepeda, Orlando; Hunt, Peter W.; Hatano, Hiroyu; Sowinski, Stefanie; Muñoz-Arias, Isa; Greene, Warner C. (2014). "Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection.". Nature. 505 (7484): 509–514. doi:10.1038/nature12940. PMC 4047036
 . PMID 24356306.
. PMID 24356306. - ↑ Doitsh G. and Greene WC. (2016) "Dissecting How CD4 T Cells Are Lost During HIV Infection.". Cell Host Microbe.
- ↑ Fink, S. L.; Cookson, B. T. (2005). "Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells". Infection and Immunity. 73 (4): 1907–16. doi:10.1128/IAI.73.4.1907-1916.2005. PMC 1087413. PMID 15784530.
- ↑ Fernandes-Alnemri, T., T; et al. (2007). "The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation". Cell Death Differ. 14 (9): 1590–604. doi:10.1038/sj.cdd.4402194. PMC 3345951. PMID 17599095. Retrieved 6 March 2011.
- ↑ Baroja-Mazo A et Al.. (2014) "The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response.". Nat Immunol..
- ↑ Franklin BS A et Al.. (2014) "The adaptor ASC has extracellular and 'prionoid' activities that propagate inflammation.". Nat Immunol..
- ↑ Cookson,B.T& Brennan, M.A.; Brennan (2001). "Pro-inflammatory programmed cell death". Trends in Microbiology. 9 (3): 113–114. doi:10.1016/S0966-842X(00)01936-3.
- 1 2 L.Duprez; et al. (2009). "Major cell death pathways at a glance". Microbes and Infection. 11 (13): 10501062. doi:10.1016/j.micinf.2009.08.013.
- ↑ M.Dagenais, A.Skeldon and M.Saleh (2012). "The inflammasome: in memory of Dr. Jurg Tschopp". Cell Death and Differentiation. 19 (1): 5–12. doi:10.1038/cdd.2011.159. PMC 3252823. PMID 22075986.
- 1 2 Karina R. Bortoluci & Ruslan Medzhitov; Medzhitov (2010). "Control of infection by pyroptosis and autophagy: role of TLR and NLR Cell". Biomedical & Life Sciences. 67 (10): 1643–1651. doi:10.1007/s00018-010-0335-5.
- ↑ Matzinger,P. (2002). "The danger model: a renewed sense of self". Science. 296 (5566): 301–305. Bibcode:2002Sci...296..301M. doi:10.1126/science.1071059. PMID 11951032.
- ↑ Sarkar, Anasuya; Hall, Mark W.; Exline, Matthew; Hart, Judy; Knatz, Nina; Gatson, Na Tosha; Wewers, Mark D. (2006). "Caspase-1 Regulates Escherichia coli Sepsis and Splenic B Cell Apoptosis Independently of Interleukin-1β and Interleukin-18". American Journal of Respiratory and Critical Care Medicine. 174 (9): 1003–10. doi:10.1164/rccm.200604-546OC. PMID 16908867.
- 1 2 3 4 Susan L. Fink,Brad T. Cookson; Cookson (2006). "Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages". Cellular Microbiology. 8 (11): 1812–1825. doi:10.1111/j.1462-5822.2006.00751.x. PMID 16824040.
- ↑ Ayala, J. M.; Yamin, T. T.; Egger, L. A.; Chin, J; Kostura, M. J.; Miller, D. K. (1994). "IL-1 beta-converting enzyme is present in monocytic cells as an inactive 45-k Da precursor". Journal of immunology (Baltimore, Md. : 1950). 153 (6): 2592–2599. PMID 8077669.
- ↑ T Kawai1 and S Akira1 (2006). "TLR signaling". Cell Death and Differentiation. 13 (5): 816–825. doi:10.1038/sj.cdd.4401850. PMID 16410796.
- 1 2 3 Franchi L, Warner N, Viani K, Nunez G; Warner; Viani; Nuñez (2009). "Function of Nod like receptors in microbial recognition and host defense". Immunol. 227 (1): 106–128. doi:10.1111/j.1600-065X.2008.00734.x. PMC 2679989. PMID 19120480.
- ↑ Suzuki, T.; et al. (2006). "Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages". PLoS Pathog. 13 (8): 816–825. doi:10.1371/journal.ppat.0030111.
- 1 2 Kufer,T.A & Sansonetti, P.J.; Sansonetti (2007). "Sensing of bacteria: NOD a lonely job". Current Opinion in Microbiology. 10 (1): 62–69. doi:10.1016/j.mib.2006.11.003. PMID 17161646.
- ↑ Y.Ogura1, F.S. Sutterwala1 & R.A. Flavell; Sutterwala; Flavell (2006). "The Inflammasome: First Line of the Immune Response to Cell Stress". Cell Press. 126 (4): 659–652. doi:10.1016/j.cell.2006.08.002.
- ↑ G. Fantuzzi & C.A. Dinarello; Dinarello (1998). "Interleukin-18 and Interleukin-1β: Two Cytokine Substrates for ICE (Caspase-1)". Journal of Clinical Immunology. 19 (1): 1–11. doi:10.1023/A:1020506300324.
- 1 2 Labbe, Katherine; Saleh, Maya (2011). The Inflammasomes. Springer Basel. pp. 17–36. ISBN 978-3-0348-0147-8.
- ↑ Nakanishi, K., Yoshimoto, T., Tsutsui, H. & Okamura, H.\; Wen; Ting (2011). "The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases". Immunol. 29 (1): 707–735. doi:10.1146/annurev-immunol-031210-101405.
- ↑ B.K. Davis, HaitaoWen & J.P.-Y. Ting; Wen; Ting (2011). "The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases". Immunol. 29 (1): 707–735. doi:10.1146/annurev-immunol-031210-101405.
- ↑ Vandanmagsar, B.; et al. (2011). "The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance". Nature Medicine. 17 (2): 179–188. doi:10.1038/nm.2279.
- ↑ T.Strowig, J.Henao-Mejia, E.Elinav & R.Flavell; Henao-Mejia; Elinav; Flavell (2012). "Inflammasomes in health and disease". Nature. 481 (7381): 278–286. Bibcode:2012Natur.481..278S. doi:10.1038/nature10759.
- ↑ B. Neven; A. Prieur; P. Quartier dit Maire (2008). "Cryopyrinopathies: update on pathogenesis and treatment". Nature Clinical Practice Rheumatology. 4 (9): 481–489. doi:10.1038/ncprheum0874.
- ↑ L.D Church; G.P Cook; M.F McDermott (2008). "Primer: inflammasomes and interleukin 1β in inflammatory disorders". Nature Clinical Practice Rheumatology. 4 (1): 34–42. doi:10.1038/ncprheum0681. PMID 18172447.
- ↑ Doitsh, Gilad; Galloway, Nicole L. K.; Geng, Xin; Yang, Zhiyuan; Monroe, Kathryn M.; Zepeda, Orlando; Hunt, Peter W.; Hatano, Hiroyu; Sowinski, Stefanie; Muñoz-Arias, Isa; Greene, Warner C. (2014). "Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection". Nature. 505 (7484): 509–14. Bibcode:2014Natur.505..509D. doi:10.1038/nature12940. PMC 4047036. PMID 24356306.
- ↑ Monroe, K. M.; Yang, Z; Johnson, J. R.; Geng, X; Doitsh, G; Krogan, N. J.; Greene, W. C. (2014). "IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV". Science. 343 (6169): 428–32. Bibcode:2014Sci...343..428M. doi:10.1126/science.1243640. PMC 3976200. PMID 24356113.
- ↑ Kuida, K; Lippke, J. A.; Ku, G; Harding, M. W.; Livingston, D. J.; Su, M. S.; Flavell, R. A. (1995). "Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme". Science. 267 (5206): 2000–3. doi:10.1126/science.7535475. PMID 7535475.
- ↑ Li, P; Allen, H; Banerjee, S; Franklin, S; Herzog, L; Johnston, C; McDowell, J; Paskind, M; Rodman, L; Salfeld, J (1995). "Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock". Cell. 80 (3): 401–11. doi:10.1016/0092-8674(95)90490-5. PMID 7859282.