Type I tyrosinemia
| Type I tyrosinemia | |
|---|---|
 | |
| Tyrosine | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E70.2 |
| ICD-9-CM | 270.2 |
| OMIM | 276700 |
| DiseasesDB | 13478 |
| eMedicine | ped/2339 |
| MeSH | D020176 |
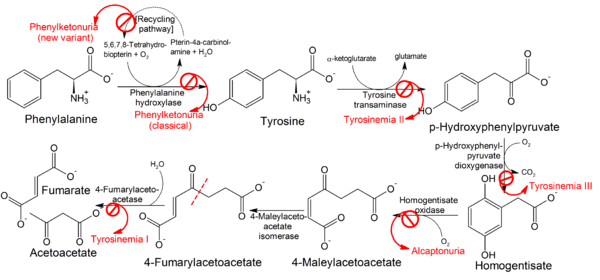
Type 1 tyrosinemia, also known as hepatorenal tyrosinemia or tyrosinosis, is the most severe form of tyrosinemia, a buildup of too much of the amino acid tyrosine in the blood and tissues due to an inability to metabolize it. It is caused by a deficiency of the enzyme fumarylacetoacetate hydrolase.
Genetics
Type 1 tyrosinemia is inherited in an autosomal recessive pattern.[1] Worldwide, type I tyrosinemia affects about 1 person in 100,000. This type of tyrosinemia is much more common in Quebec, Canada. The overall incidence in Quebec is about 1 in 16,000 individuals. In the Saguenay-Lac-Saint-Jean region of Quebec, type 1 tyrosinemia affects 1 person in 1,846.[2] The carrier rate has been estimated to be between 1 in 20 and 1 in 31.[3]
Pathophysiology
Fumarylacetoacetate hydrolase catalyzes the final step in the degradation of tyrosine - fumarylacetoacetate to fumarate, acetoacetate and succinate. Fumarylacetoacetate accumulates in hepatocytes and proximal renal tubal cells and causes oxidative damage and DNA damage leading to cell death and dysfunctional gene expression which alters metabolic processes like protein synthesis and gluconeogenesis. The increase in fumarylacetoacetate inhibits previous steps in tyrosine degradation leading to an accumulation of tyrosine in the body. Tyrosine is not directly toxic to the liver or kidneys but causes dermatologic and neurodevelopmental problems.
Signs and symptoms
Type 1 tyrosinemia typically presents in infancy as failure to thrive and hepatomegaly. The primary effects are progressive liver and kidney dysfunction. The liver disease causes cirrhosis, conjugated hyperbilirubinemia, elevated AFP, hypoglycemia and coagulation abnormalities. This can lead to jaundice, ascites and hemorrhage. There is also an increased risk of hepatocellular carcinoma. The kidney dysfunction presents as Fanconi syndrome: Renal tubular acidosis, hypophosphatemia and aminoaciduria. Cardiomyopathy, neurologic and dermatologic manifestations are also possible. The urine has an odor of cabbage or rancid butter.[4]
Treatment
The primary treatment for type 1 tyrosinemia is nitisinone (Orfadin) and restriction of tyrosine in the diet.[1] Nitisinone inhibits the conversion of 4-OH phenylpyruvate to homogentisic acid by 4-Hydroxyphenylpyruvate dioxygenase, the second step in tyrosine degradation. By inhibiting this enzyme, the accumulation of the fumarylacetoacetate is prevented.[5] Previously, liver transplantation was the primary treatment option and is still used in patients in whom nitisinone fails.
References
- 1 2 National Organization for Rare Disorders. Physician’s Guide to Tyrosinemia Type 1
- ↑ Grompe M, St-Louis M, Demers SI, al-Dhalimy M, Leclerc B, Tanguay RM (1994). "A single mutation of the fumarylacetoacetate hydrolase gene in French Canadians with hereditary tyrosinemia type I". N. Engl. J. Med. 331 (6): 353–7. doi:10.1056/NEJM199408113310603. PMID 8028615.
- ↑ Laberge, C.; L. Dallaire (October 28, 1967). "Genetic aspects of tyrosinemia in the Chicoutimi region". Can Med Assoc J. 97 (18): 1099–1101. PMC 1923580
 . PMID 6057677.
. PMID 6057677. - ↑ Enns GM, Packman S (2001). "Diagnosing Inborn Errors of Metabolism in the Newborn: Clinical Features" (PDF). NeoReviews. 2 (8): e183–e191. doi:10.1542/neo.2-8-e183. ISSN 1526-9906.
- ↑ Lock EA et al. From toxicological problem to therapeutic use: the discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), its toxicology and development as a drug. J Inherit Metab Dis. 1998 Aug;21(5):498-506. PMID 9728330