Transthyretin-related hereditary amyloidosis
| Familial amyloid polyneuropathy | |
|---|---|
| Classification and external resources | |
| OMIM | 105210 |
| eMedicine | article/335301 |
Familial amyloid polyneuropathy (FAP), also called transthyretin-related hereditary amyloidosis, transthyretin amyloidosis abbreviated also as ATTR ( hereditary form), or Corino de Andrade's disease,[1] is an autosomal dominant[2] neurodegenerative disease. It is a form of amyloidosis, and was first identified and described by Portuguese neurologist Mário Corino da Costa Andrade, in 1952.[3] FAP is distinct from senile systemic amyloidosis (SSA), which is not inherited, and which was determined to be the primary cause of death for 70% of supercentenarians who have been autopsied.[4]
FAP can be ameliorated by liver transplantation.
Characteristics
Usually manifesting itself between 20 and 40 years of age, it is characterized by pain, paresthesia, muscular weakness and autonomic dysfunction. In its terminal state, the kidneys and the heart are affected. FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
Cause and genetics
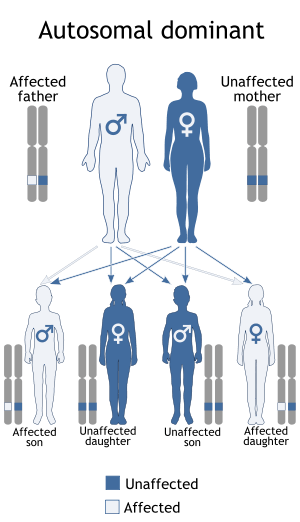
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver. The transthyretin protein is a tetramer. The tetramer has to dissociate into misfolded monomers to aggregate into a variety of structures including amyloid fibrils. Because most patients are heterozygotes, they deposit both mutant and wild type TTR subnits.
FAP is inherited in an autosomal dominant manner.[2] This means that the defective gene responsible for the disorder is located on an autosome (chromosome 18 is an autosome), and only one copy of the defective gene is sufficient to cause the disorder, when inherited from a parent who has the disorder.
Prognosis
In the absence of a liver transplant, FAP is invariably fatal, usually within a decade. The disadvantage of liver transplantation is that approximately 10% of the subjects die from the procedure or complications resulting from the procedure, which is a form of gene therapy wherein the liver expressing wild type and mutant TTR is replaced by a liver only expressing wild type TTR. Moreover, transplanted patients must take immune suppressants (drugs) for the remainder of their life, which can lead to additional complications. In late 2011, the European Medicines Agency approved the transthyretin kinetic stabilizer Tafamidis or Vyndaqel discovered by Jeffery W. Kelly and developed by FoldRx pharmaceuticals (acquired by Pfizer in 2010) for the treatment of FAP based on clinical trial data. Tafamidis (20 mg once daily) slowed the progression of FAP over a 36-month period and importantly reversed the weight loss and muscle wasting associated with disease progression.
Epidemiology
This disease is endemic in Portuguese locations Póvoa de Varzim and Vila do Conde (Caxinas), with more than 1000 affected people, coming from about 500 families, where 70% of the people develop the illness. ll the analysed Portuguese families presented the same haplotype (haplotype I) associated with the Met 30 mutation. In northern Sweden, more specifically Piteå, Skellefteå and Umeå, 1.5% of the population has the mutated gene. There are many other populations in the world who exhibit the illness after having developed it independently.
Regulatory Agency Approved Treatments
The drug tafamidis[6] has completed a phase II/III 18-month-long placebo controlled clinical trial[7] and these results in combination with an 18-month follow-on study demonstrated that Tafamidis or Vyndaqel slowed progression of FAP, particularly when administered to patients early in the course of FAP.[8] This drug is now approved by the European Medicines Agency.
The US Food and Drug Administration's Peripheral and Central Nervous System Drugs Advisory Committee rejected the drug in June 2012, in a 13-4 vote.[9] The committee stated that there was not enough evidence supporting efficacy of the drug, and requested additional clinical trials.[10]
References
- 1 2 Online Mendelian Inheritance in Man (OMIM) 105210
- 1 2 Ando, Y.; Ueda, M. (May 2008). "Novel methods for detecting amyloidogenic proteins in transthyretin related amyloidosis". Frontiers in Bioscience. 13: 5548–5558. PMID 18508604.
- ↑ Andrade C (September 1952). "A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves". Brain. 75 (3): 408–27. doi:10.1093/brain/75.3.408. PMID 12978172.
- ↑ Coles LS, Young RD (2012). "Supercentenarians and transthyretin amyloidosis: the next frontier of human life extension". PREVENTATIVE MEDICINE. 54 (Suppl): s9–s11. doi:10.1016/j.ypmed.2012.03.003. PMID 22579241.
- ↑ Online Mendelian Inheritance in Man (OMIM) 176300
- ↑ Tafamidis structure
- ↑ Safety and Efficacy Study of Fx-1006A in Patients With Familial Amyloidosis – ClinicalTrials.gov ref: NCT00409175
- ↑ http://protomag.ticsnetwork.com/statics/MGH_SP10_Protein_fold_F2.pdf
- ↑ http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM304830.pdf
- ↑ http://www.bioworld.com/content/fda-delivers-crl-pfizers-rare-disease-drug-vyndaqel-0
External links
- GeneReviews/NIH/NCBI/UW entry on Familial Transthyretin Amyloidosis
- Stanford University Amyloid Center