Sirolimus
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Rapamune |
| License data | |
| Pregnancy category | |
| Routes of administration | Oral |
| ATC code | L04AA10 (WHO) S01XA23 (WHO) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 14% (oral solution), lower with high-fat meals; 18% (tablet), higher with high-fat meals[1] |
| Protein binding | 92% |
| Metabolism | Hepatic |
| Biological half-life | 57–63 hours[2] |
| Excretion | Mostly faecal |
| Identifiers | |
| |
| Synonyms | Rapamycin |
| CAS Number |
53123-88-9 |
| PubChem (CID) | 5284616 |
| DrugBank |
DB00877 |
| ChemSpider |
10482078 |
| UNII |
W36ZG6FT64 |
| KEGG |
D00753 |
| ChEBI |
CHEBI:9168 |
| ChEMBL |
CHEMBL413 |
| PDB ligand ID | RAP (PDBe, RCSB PDB) |
| ECHA InfoCard | 100.107.147 |
| Chemical and physical data | |
| Formula | C51H79NO13 |
| Molar mass | 914.172 g/mol |
| 3D model (Jmol) | Interactive image |
| Solubility in water | 0.0026 [3] mg/mL (20 °C) |
| |
| |
| (verify) | |
Sirolimus (INN/USAN), also known as rapamycin, is a macrolide compound that is used to coat coronary stents, prevent organ transplant rejection and to treat a rare lung disease called lymphangioleiomyomatosis.[4][5][6] It has immunosuppressant functions in humans and is especially useful in preventing the rejection of kidney transplants. It inhibits activation of T cells and B cells by reducing the production of interleukin-2 (IL-2).
It is produced by the bacterium Streptomyces hygroscopicus and was isolated for the first time in 1972 by Suren Sehgal and colleagues from samples of Streptomyces hygroscopicus found on Easter Island.[7][8] The compound was originally named rapamycin after the native name of the island, Rapa Nui.[5] Sirolimus was initially developed as an antifungal agent. However, this use was abandoned when it was discovered to have potent immunosuppressive and antiproliferative properties due to its ability to inhibit mTOR. It was approved by the US Food and Drug Administration in September 1999 and is marketed under the trade name Rapamune by Pfizer (formerly by Wyeth).
Medical uses
Sirolimus is indicated for the prevention of organ transplant rejection and for the treatment of lymphangioleiomyomatosis (LAM).[4]
Prevention of transplant rejection
The chief advantage sirolimus has over calcineurin inhibitors is its low toxicity toward kidneys. Transplant patients maintained on calcineurin inhibitors long-term tend to develop impaired kidney function or even chronic renal failure; this can be avoided by using sirolimus instead. It is particularly advantageous in patients with kidney transplants for hemolytic-uremic syndrome, as this disease is likely to recur in the transplanted kidney if a calcineurin-inhibitor is used. However, on 7 October 2008, the FDA approved safety labeling revisions for sirolimus to warn of the risk for decreased renal function associated with its use.[9][10] In 2009, the FDA notified healthcare professionals that a clinical trial conducted by Wyeth showed an increased mortality in stable liver transplant patients after switching from a calcineurin inhibitor-based immunosuppressive regimen to sirolimus.[11]
Sirolimus can also be used alone, or in conjunction with a calcineurin inhibitor (such as tacrolimus), and/or mycophenolate mofetil, to provide steroid-free immunosuppression regimens. Impaired wound healing and thrombocytopenia are a possible side effects of sirolimus; therefore, some transplant centers prefer not to use it immediately after the transplant operation, but instead administer it only after a period of weeks or months. Its optimal role in immunosuppression has not yet been determined, and it remains the subject of a number of ongoing clinical trials.[12]
Lymphangioleiomyomatosis
On May 28, 2015, the FDA approved sirolimus to treat lymphangioleiomyomatosis (LAM), a rare, progressive lung disease that primarily affects women of childbearing age. This made sirolimus the first drug approved to treat this disease.[13] LAM involves lung tissue infiltration with smooth muscle-like cells with mutations of the tuberous sclerosis complex gene (TSC2). Loss of TSC2 gene function activates the mTOR signaling pathway, resulting in the release of lymphangiogenic growth factors. Sirolimus blocks this pathway.[4]
The safety and efficacy of sirolimus treatment of LAM were investigated in clinical trials that compared sirolimus treatment with a placebo group in 89 patients for 12 months. The patients were observed for 12 months after the treatment had ended. The most commonly reported side effect of sirolimus treatment of LAM were mouth and lip ulcers, diarrhea, abdominal pain, nausea, sore throat, acne, chest pain, leg swelling, upper respiratory tract infection, headache, dizziness, muscle pain and elevated cholesterol. Serious side effects including hypersensitivity and swelling (edema) have been observed in renal transplant patients.[13]
While sirolimus was considered for treatment of LAM, it received orphan product designation status because LAM is a rare condition. Development for the product was partially supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and/or effectiveness of products for use in rare diseases or conditions.[13]
The safety of LAM treatment by sirolimus in patients younger than 18 years old has not been tested.[4]
Coronary stent coating
The antiproliferative effect of sirolimus has also been used in conjunction with coronary stents to prevent restenosis in coronary arteries following balloon angioplasty. The sirolimus is formulated in a polymer coating that affords controlled release through the healing period following coronary intervention. Several large clinical studies have demonstrated lower restenosis rates in patients treated with sirolimus-eluting stents when compared to bare-metal stents, resulting in fewer repeat procedures. A sirolimus-eluting coronary stent was marketed by Cordis, a division of Johnson & Johnson, under the tradename Cypher.[6] However, this kind of stent may also increase the risk of vascular thrombosis.[14]
Contraindications
Sirolimus is contraindicated in individuals with a known hypersensitivity to the drug.[4]
Adverse effects
The most common adverse reactions (≥30% occurrence, leading to a 5% treatment discontinuation rate) observed with sirolimus in clinical studies of organ rejection prophylaxis in individuals with kidney transplants include: peripheral edema, hypercholesterolemia, abdominal pain, headache, nausea, diarrhea, pain, constipation, hypertriglyceridemia, hypertension, increased creatinine, fever, urinary tract infection, anemia, arthralgia, and thrombocytopenia.[4]
The most common adverse reactions (≥20% occurrence, leading to a 11% treatment discontinuation rate) observed with sirolimus in clinical studies for the treatment of lymphangioleiomyomatosis are: peripheral edema, hypercholesterolemia, abdominal pain, headache, nausea, diarrhea, chest pain, stomatitis, nasopharyngitis, acne, upper respiratory tract infection, dizziness, and myalgia.[4]
The following adverse effects occurred in 3–20% of individuals taking sirolimus for organ rejection prophylaxis following a kidney transplant:[4]
| System | Adverse effects |
|---|---|
| Body as a Whole | Sepsis, lymphocele, herpes zoster infection, herpes simplex infection |
| Cardiovascular | Venous thromboembolism (pulmonary embolism and deep venous thrombosis), rapid heart rate |
| Digestive | Stomatitis |
| Hematologic/Lymphatic | Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (TTP/HUS), leukopenia |
| Metabolic | Abnormal healing, increased lactic dehydrogenase (LDH), hypokalemia, diabetes |
| Musculoskeletal | Bone necrosis |
| Respiratory | Pneumonia, epistaxis |
| Skin | Melanoma, squamous cell carcinoma, basal cell carcinoma |
| Urogenital | Pyelonephritis, ovarian cysts, menstrual disorders (amenorrhea and menorrhagia) |
Diabetes-like symptoms
While sirolimus inhibition of mTORC1 appears to mediate the drug's benefits, it also inhibits mTORC2, which results in diabetes-like symptoms. This includes decreased glucose tolerance and insensitivity to insulin.[15] Sirolimus treatment may additionally increase the risk of type 2 diabetes.[16] In mouse studies, these symptoms can be avoided through the use of alternate dosing regimens or analogs such as everolimus or temsirolimus.[17]
Lung toxicity
Lung toxicity is a serious complication associated with sirolimus therapy,[18][19][20][21][22][23][24] especially in the case of lung transplants.[25] The mechanism of the interstitial pneumonitis caused by sirolimus and other macrolide MTOR inhibitors is unclear, and may have nothing to do with the mTOR pathway.[26][27][28] The interstitial pneumonitis is not dose-dependent, but is more common in patients with underlying lung disease.[18][29]
Lowered effectiveness of immune system
There have been warnings about the use of sirolimus in transplants, where it may increase mortality due to an increased risk of infections.[4][30]
Cancer risk
According to FDA prescribing information, sirolimus may increase an individual's risk for contracting skin cancers from exposure to sunlight or UV radiation, and risk of developing lymphoma.[4]
Impaired wound healing
Individuals taking sirolimus are at increased risk of experiencing impaired or delayed wound healing, particularly if they have a high body mass index (i.e., a BMI of ≥30 kg/m2).[4]
Interactions
Sirolimus is metabolized by the CYP3A4 enzyme and is a substrate of the P-glycoprotein (P-gp) efflux pump;[4] hence, inhibitors of either protein may increase sirolimus concentrations in blood plasma, whereas inducers of CYP3A4 and P-gp may decrease sirolimus concentrations in blood plasma.[4]
Pharmacology
Pharmacodynamics
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits IL-2 and other cytokines receptor-dependent signal transduction mechanisms, via action on mTOR, and thereby blocks activation of T and B cells. Tacrolimus and cyclosporine inhibit the secretion of IL-2, by inhibiting calcineurin.[12]
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12 (FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex, which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits the mTOR (mechanistic (formerly mammalian) Target Of Rapamycin, rapamycin being another name for sirolimus) pathway by directly binding to mTOR Complex 1 (mTORC1).[12]
mTOR has also been called FRAP (FKBP-rapamycin-associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.[12]
Pharmacokinetics
Sirolimus is metabolized by the CYP3A4 enzyme and is a substrate of the P-glycoprotein (P-gp) efflux pump.[4] It has an elimination half-life of 57–63 hours.[2]
The absorption of sirolimus into the blood stream from the intestine varies widely between patients, with some patients having up to eight times more exposure than others for the same dose. Drug levels are, therefore, taken to make sure patients get the right dose for their condition.[12] This is determined by taking a blood sample before the next dose, which gives the trough level. However, good correlation is noted between trough concentration levels and drug exposure, known as area under the concentration-time curve, for both sirolimus (SRL) and tacrolimus (TAC) (SRL: r2 = 0.83; TAC: r2 = 0.82), so only one level need be taken to know its pharmacokinetic (PK) profile. PK profiles of SRL and of TAC are unaltered by simultaneous administration. Dose-corrected drug exposure of TAC correlates with SRL (r2 = 0.8), so patients have similar bioavailability of both.[31]
Chemistry
Sirolimus is a natural product and macrocyclic lactone.[2]
Biosynthesis
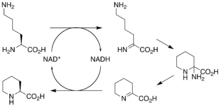
The biosynthesis of the rapamycin core is accomplished by a type I polyketide synthase (PKS) in conjunction with a nonribosomal peptide synthetase (NRPS). The domains responsible for the biosynthesis of the linear polyketide of rapamycin are organized into three multienzymes, RapA, RapB, and RapC, which contain a total of 14 modules (figure 1). The three multienzymes are organized such that the first four modules of polyketide chain elongation are in RapA, the following six modules for continued elongation are in RapB, and the final four modules to complete the biosynthesis of the linear polyketide are in RapC.[32] Then, the linear polyketide is modified by the NRPS, RapP, which attaches L-pipecolate to the terminal end of the polyketide, and then cyclizes the molecule, yielding the unbound product, prerapamycin.[33]
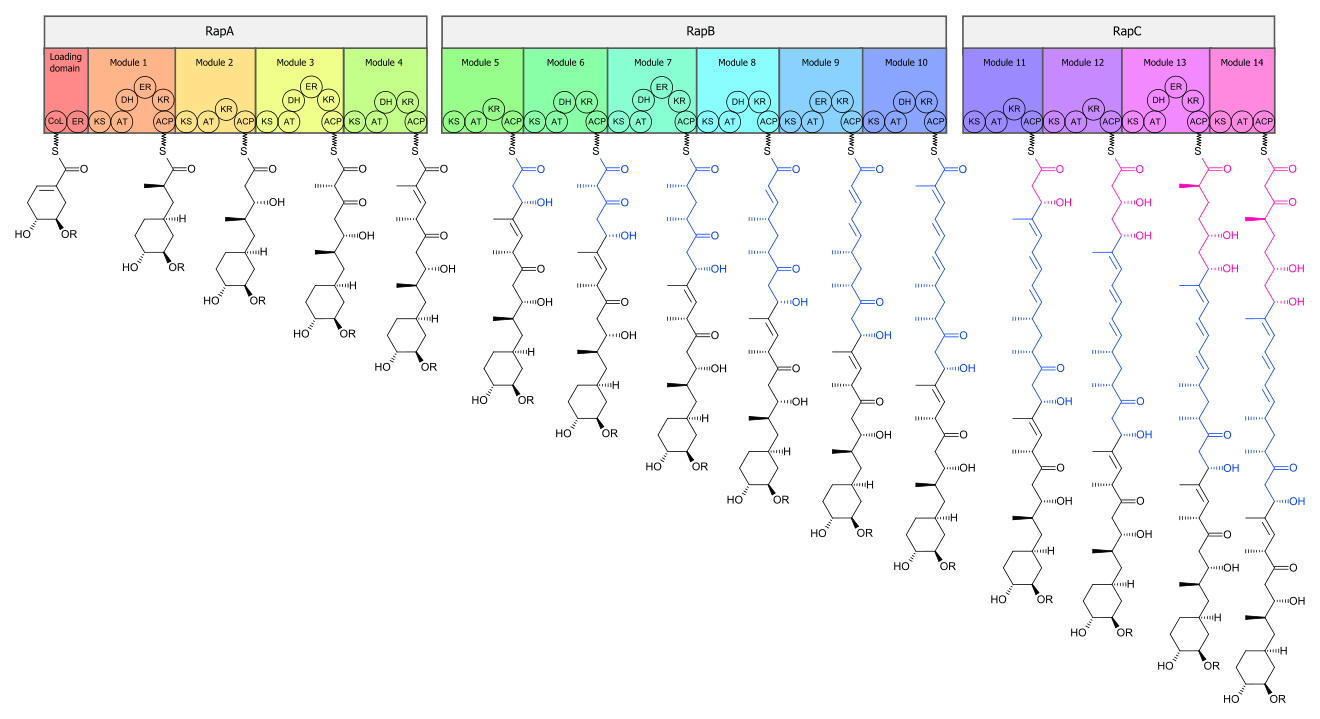
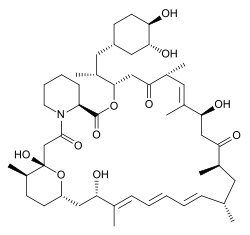
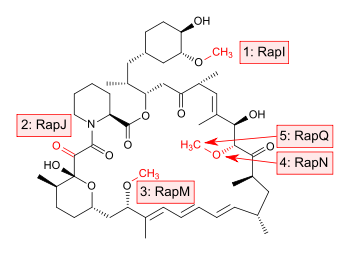
The core macrocycle, prerapamycin (figure 2), is then modified (figure 3) by an additional five enzymes, which lead to the final product, rapamycin. First, the core macrocycle is modified by RapI, SAM-dependent O-methyltransferase (MTase), which O-methylates at C39. Next, a carbonyl is installed at C9 by RapJ, a cytochrome P-450 monooxygenases (P-450). Then, RapM, another MTase, O-methylates at C16. Finally, RapN, another P-450, installs a hydroxyl at C27 immediately followed by O-methylation by Rap Q, a distinct MTase, at C27 to yield rapamycin.[34]
The biosynthetic genes responsible for rapamycin synthesis have been identified. As expected, three extremely large open reading frames (ORF's) designated as rapA, rapB, and rapC encode for three extremely large and complex multienzymes, RapA, RapB, and RapC, respectively.[32] The gene rapL has been established to code for a NAD+-dependent lysine cycloamidase, which converts L-lysine to L-pipecolic acid (figure 4) for incorporation at the end of the polyketide.[35][36] The gene rapP, which is embedded between the PKS genes and translationally coupled to rapC, encodes for an additional enzyme, an NPRS responsible for incorporating L-pipecolic acid, chain termination and cyclization of prerapamycin. In addition, genes rapI, rapJ, rapM, rapN, rapO, and rapQ have been identified as coding for tailoring enzymes that modify the macrocyclic core to give rapamycin (figure 3). Finally, rapG and rapH have been identified to code for enzymes that have a positive regulatory role in the preparation of rapamycin through the control of rapamycin PKS gene expression.[37] Biosynthesis of this 31-membered macrocycle begins as the loading domain is primed with the starter unit, 4,5-dihydroxocyclohex-1-ene-carboxylic acid, which is derived from the shikimate pathway.[32] Note that the cyclohexane ring of the starting unit is reduced during the transfer to module 1. The starting unit is then modified by a series of Claisen condensations with malonyl or methylmalonyl substrates, which are attached to an acyl carrier protein (ACP) and extend the polyketide by two carbons each. After each successive condensation, the growing polyketide is further modified according to enzymatic domains that are present to reduce and dehydrate it, thereby introducing the diversity of functionalities observed in rapamycin (figure 1). Once the linear polyketide is complete, L-pipecolic acid, which is synthesized by a lysine cycloamidase from an L-lysine, is added to the terminal end of the polyketide by an NRPS. Then, the NSPS cyclizes the polyketide, giving prerapamycin, the first enzyme-free product. The macrocyclic core is then customized by a series of post-PKS enzymes through methylations by MTases and oxidations by P-450s to yield rapamycin.
Research

Cancer
The antiproliferative effects of sirolimus may have a role in treating cancer. When dosed appropriately, sirolimus can enhance the immune response to tumor targeting[38] or otherwise promote tumor regression in clinical trials.[39] Sirolimus seems to lower the cancer risk in some transplant patients.[40]
Sirolimus was shown to inhibit the progression of dermal Kaposi's sarcoma in patients with renal transplants. Other mTOR inhibitors, such as temsirolimus (CCI-779) or everolimus (RAD001), are being tested for use in cancers such as glioblastoma multiforme and mantle cell lymphoma. However, these drugs have a higher rate of fatal adverse events in cancer patients than control drugs.[41]
A combination therapy of doxorubicin and sirolimus has been shown to drive AKT-positive lymphomas into remission in mice. Akt signalling promotes cell survival in Akt-positive lymphomas and acts to prevent the cytotoxic effects of chemotherapy drugs, such as doxorubicin or cyclophosphamide. Sirolimus blocks Akt signalling and the cells lose their resistance to the chemotherapy. Bcl-2-positive lymphomas were completely resistant to the therapy; eIF4E-expressing lymphomas are not sensitive to sirolimus.[42][43][44][45]
Tuberous sclerosis complex
Sirolimus also shows promise in treating tuberous sclerosis complex (TSC), a congenital disorder that leaves sufferers prone to benign tumor growth in the brain, heart, kidneys, skin, and other organs. After several studies conclusively linked mTOR inhibitors to remission in TSC tumors, specifically subependymal giant-cell astrocytomas in children and angiomyolipomas in adults, many US doctors began prescribing sirolimus (Wyeth's Rapamune) and everolimus (Novartis's RAD001) to TSC patients off-label. Numerous clinical trials using both rapamycin analogs, involving both children and adults with TSC, are underway in the United States.[46]
Most studies thus far have noted that tumors often regrew when treatment stopped.
Facial angiofibromas occur in 80% of patients with TSC, and the condition is very disfiguring. A retrospective review of English-language medical publications reporting on topical sirolimus treatment of facial angiofibromas found sixteen separate studies with positive patient outcomes after using the drug. The reports involved a total of 84 patients, and improvement was observed in 94% of subjects, especially if treatment began during the early stages of the disease. Sirolimus treatment was applied in several different formulations (ointment, gel, solution, and cream), ranging from 0.003 to 1% concentrations. Reported adverse effects included one case of perioral dermatitis, one case of cephalea, and four cases of irritation.[47]
Effects on longevity
Rapamycin was first shown to extend lifespan in eukaryotes in 2006 by Powers et al. who showed a dose-responsive effect of rapamycin on lifespan extension in yeast cells.[48] Building on this and other work, in a 2009 study, the lifespans of mice fed rapamycin were increased between 28 and 38% from the beginning of treatment, or 9 to 14% in total increased maximum lifespan. Of particular note, the treatment began in mice aged 20 months, the equivalent of 60 human years.[49] Rapamycin has subsequently been shown to extend mouse lifespan in several separate experiments,[50][51] and is now being tested for this purpose in nonhuman primates (the marmoset monkey),[52] and with an ongoing attempt to organize a study in dogs.[53] The Dog Aging Project is funded by pet owners.[54]
Because rapamycin at high doses can suppress the immune system, people taking rapamycin for transplant or cancer therapy are warned of potentially greater susceptibility to dangerous infections.[4] Yet paradoxically, rapamycin was shown to enhance the ability of aging mice to mount an immune response to a vaccine against tuberculosis. A similar immunological "rejuvenating" effect was later documented in elderly humans administered a rapamycin analog prior to influenza vaccination), further fueling optimism for the potential of mTOR as a target for anti-aging drugs for humans. It is not known whether rapamycin will have similar lifespan-lengthening effects in humans, and the authors of one study caution that the drug should not be used by the general population for this purpose.[55] It is hypothesized that some dietary regimes, like caloric restriction and methionine restriction, cause lifespan extension by decreasing mTOR activity.[56] It is believed that this is achieved by limiting the essential amino acid leucine. Leucine is a potent activator of mTORC1.[57] The administration of leucine into the rat brain has been shown to decrease food intake and body weight via activation of the mTOR pathway.[58]
According to the free radical theory of aging,[59] reactive oxygen species cause damage of mitochondrial proteins and decrease of ATP production. Subsequently, via ATP sensitive AMPK, the mTOR pathway is inhibited and ATP consuming protein synthesis is downregulated, since mTORC1 initiates a phosphorylation cascade activating the ribosome.[60] Hence, the proportion of damaged proteins is enhanced. Moreover, disruption of mTORC1 directly inhibits mitochondrial respiration.[61] These positive feedbacks on the aging process are counteracted by protective mechanisms: Decreased mTOR activity (among other factors) upregulates glycolysis[61] and removal of dysfunctional cellular components via autophagy.[59]
Tuberous sclerosis in mice, with possible relevance to autism
In a study of sirolimus as a treatment for tuberous sclerosis, researchers observed improvements in TSC symptoms which overlap with autism. The researchers discovered that sirolimus regulates one of the same proteins the TSC gene does, but in different parts of the body. They decided to treat mice three to six months old (adulthood in mice lifespans); this increased the TSC mice's intellect to about that of normal mice in as little as three days.[62]
Alzheimer's in mice
Sirolimus reduced brain lesions and prevented the decline of performance in the water maze in mice with a mouse model of Alzheimer's.[63] Recent studies have observed a protective effect against Alzheimer's Disease in preventing cognitive deficits and reducing amyloid-β levels in mouse models. [64]
Muscular dystrophy in mice
Researchers at Washington University School of Medicine in St. Louis observed that administration of nanoparticles coated in sirolimus increases grip strength by 30% and significantly increases cardiac function in mice. The nanoparticles consist of perfluorocarbon cores and are 200 nm in diameter. The nanoparticles accumulate in areas of inflammation, in this case muscle, where they release a small dose of sirolimus. This suppresses the immune system and promotes autophagy.[65]
Systemic lupus erythematosus in mice and humans
Sirolimus improves disease activity and dependence on prednisone in systemic lupus erythematosus (SLE) patients resistant or intolerant to immunosuppressant medications. Sirolimus acts through blocking the activation of its molecular target, the mechanistic target of rapamycin complex 1 (mTORC1). The activation of mTORC1, which is associated with suppression of mTORC2, results in the expansion of proinflammatory CD4-CD8- double-negative (DN) T lymphocytes. These DN T cells produce inflammatory cytokines, interleukin-4 (IL-4) and interleukin-17, and they exhibit predisposition to proinflammatory cell death through necrosis. Increased IL-4 production is responsible for activation of autoantibody-producing B lymphocytes in SLE.[66][67][68][69][70] Sirolimus also blocks disease in lupus-prone mice by reversing the activation of mTORC1.[71] Prospective clinical trial in SLE patients with sirolimus is ongoing.
Other afflictions
Studies in vitro, in mice, and in humans suggest sirolimus inhibits HIV replication through different mechanisms, including downregulation of the coreceptor CCR5[72] and the induction of autophagy.[73]
In addition, sirolimus is currently being assessed as a therapeutic option for autosomal-dominant polycystic kidney disease (ADPKD). Case reports indicate sirolimus can reduce kidney volume and delay the loss of renal function in patients with ADPKD.[74]
Sirolimus has also been used in preliminary research to combat progeria, a rare disorder that causes individuals to age at an exceedingly rapid pace, leading to an extremely compromised cell-damage repair capacity and typically resulting in death in the early teenage years due to causes which are generally associated with old age such as heart disease or stroke.[75]
Applications in biology research
Rapamycin is used in biology research as an agent for chemically induced dimerization.[76] In this application, rapamycin is added to cells expressing two fusion constructs, one of which contains the rapamycin-binding FRB domain from mTOR and the other of which contains an FKBP domain. Each fusion protein also contains additional domains that are brought into proximity when rapamycin induces binding of FRB and FKBP. In this way, rapamycin can be used to control and study protein localization and interactions.
References
- ↑ Buck, Marcia L. (2006). "Immunosuppression With Sirolimus After Solid Organ Transplantation in Children". Pediatric Pharmacotherapy. 12 (2).
- 1 2 3 4 "Rapamycin". PubChem Compound. National Center for Biotechnology Information. Retrieved 1 August 2016.
- ↑ Simamora, P; Alvarez, JM; Yalkowsky, SH (1 February 2001). "Solubilization of rapamycin". International journal of pharmaceutics. 213 (1–2): 25–9. doi:10.1016/s0378-5173(00)00617-7. PMID 11165091.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 "Rapamune Prescribing Information" (PDF). United States Food and Drug Administration. Wyeth Pharmaceuticals, Inc. May 2015. Retrieved 28 May 2016.
- 1 2 Vézina C, Kudelski A, Sehgal SN (October 1975). "Rapamycin (AY-22,989), a new antifungal antibiotic". J. Antibiot. 28 (10): 721–6. doi:10.7164/antibiotics.28.721. PMID 1102508.
- 1 2 "Cypher Sirolimus-eluting Coronary Stent". Cypher Stent. Retrieved 1 April 2008.
- ↑ Seto, Belinda (2012). "Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer". Clinical and Translational Medicine. 1 (1): 29. doi:10.1186/2001-1326-1-29.
- ↑ Pritchard DI (2005). "Sourcing a chemical succession for cyclosporin from parasites and human pathogens". Drug Discovery Today. 10 (10): 688–691. doi:10.1016/S1359-6446(05)03395-7. PMID 15896681.
- ↑ Li, Jie Jack; Corey, E. J. (2013-04-03). Drug Discovery: Practices, Processes, and Perspectives. John Wiley & Sons. ISBN 9781118354469.
- ↑ Koprowski, Gene (2012-02-07). Nanotechnology in Medicine: Emerging Applications. Momentum Press. ISBN 9781606502501.
- ↑ "Sirolimus (marketed as Rapamune) Safety". FDA.gov. U.S. Food and Drug Administration. 11 June 2009. Retrieved 1 Aug 2016.
- 1 2 3 4 5 Mukherjee, Sandeep; Mukherjee, Urmila (2009-01-01). "A Comprehensive Review of Immunosuppression Used for Liver Transplantation". Journal of Transplantation. 2009: 1–20. doi:10.1155/2009/701464. ISSN 2090-0007. PMC 2809333
 . PMID 20130772.
. PMID 20130772. - 1 2 3 Pahon, Eric (May 28, 2015). "FDA approves Rapamune to treat LAM, a very rare lung disease". FDA.gov. U.S. Food and Drug Administration. Retrieved 1 Aug 2016.
- ↑ Shuchman M (2006). "Trading restenosis for thrombosis? New questions about drug-eluting stents". N Engl J Med. 355 (19): 1949–52. doi:10.1056/NEJMp068234. PMID 17093244.
- ↑ Lamming DW, Ye L, Katajisto P; Ye; Katajisto; Goncalves; Saitoh; Stevens; Davis; Salmon; Richardson; Ahima; Guertin; Sabatini; Baur; et al. (March 2012). "Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity". Science. 335 (6076): 1638–43. Bibcode:2012Sci...335.1638L. doi:10.1126/science.1215135. PMC 3324089. PMID 22461615.
- ↑ Johnston, Olwyn; Rose, Caren L.; Webster, Angela C.; Gill, John S. (1 July 2008). "Sirolimus is associated with new-onset diabetes in kidney transplant recipients". Journal of the American Society of Nephrology: JASN. 19 (7): 1411–1418. doi:10.1681/ASN.2007111202. ISSN 1533-3450. PMC 2440303. PMID 18385422.
- ↑ Arriola Apelo, Sebastian I.; Neuman, Joshua C.; Baar, Emma L.; Syed, Faizan A.; Cummings, Nicole E.; Brar, Harpreet K.; Pumper, Cassidy P.; Kimple, Michelle E.; Lamming, Dudley W. (13 October 2015). "Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system". Aging Cell. 15: 28–38. doi:10.1111/acel.12405. ISSN 1474-9726. PMC 4717280. PMID 26463117.
- 1 2 Chhajed PN, Dickenmann M, Bubendorf L, Mayr M, Steiger J, Tamm M (2006). "Patterns of pulmonary complications associated with sirolimus". Respiration. 73 (3): 367–74. doi:10.1159/000087945. PMID 16127266.
- ↑ Morelon E, Stern M, Israël-Biet D, et al. (September 2001). "Characteristics of sirolimus-associated interstitial pneumonitis in renal transplant patients". Transplantation. 72 (5): 787–90. doi:10.1097/00007890-200109150-00008. PMID 11571438.
- ↑ Filippone EJ, Carson JM, Beckford RA, et al. (September 2011). "Sirolimus-induced pneumonitis complicated by pentamidine-induced phospholipidosis in a renal transplant recipient: a case report". Transplant. Proc. 43 (7): 2792–7. doi:10.1016/j.transproceed.2011.06.060. PMID 21911165.
- ↑ Pham PT, Pham PC, Danovitch GM, et al. (April 2004). "Sirolimus-associated pulmonary toxicity". Transplantation. 77 (8): 1215–20. doi:10.1097/01.TP.0000118413.92211.B6. PMID 15114088.
- ↑ Mingos MA, Kane GC (December 2005). "Sirolimus-induced interstitial pneumonitis in a renal transplant patient" (PDF). Respir Care. 50 (12): 1659–61. PMID 16318648.
- ↑ Das BB, Shoemaker L, Subramanian S, Johnsrude C, Recto M, Austin EH (March 2007). "Acute sirolimus pulmonary toxicity in an infant heart transplant recipient: case report and literature review". J. Heart Lung Transplant. 26 (3): 296–8. doi:10.1016/j.healun.2006.12.004. PMID 17346635.
- ↑ Delgado JF, Torres J, José Ruiz-Cano M, et al. (September 2006). "Sirolimus-associated interstitial pneumonitis in 3 heart transplant recipients". J. Heart Lung Transplant. 25 (9): 1171–4. doi:10.1016/j.healun.2006.05.013. PMID 16962483.
- ↑ McWilliams TJ, Levvey BJ, Russell PA, Milne DG, Snell GI (February 2003). "Interstitial pneumonitis associated with sirolimus: a dilemma for lung transplantation". J. Heart Lung Transplant. 22 (2): 210–3. doi:10.1016/S1053-2498(02)00564-8. PMID 12581772.
- ↑ Aparicio G, Calvo MB, Medina V, et al. (August 2009). "Comprehensive lung injury pathology induced by mTOR inhibitors". Clin Transl Oncol. 11 (8): 499–510. doi:10.1007/s12094-009-0394-y. PMID 19661024.
- ↑ Paris A, Goupil F, Kernaonet E, et al. (January 2012). "[Drug-induced pneumonitis due to sirolimus: an interaction with atorvastatin?]". Rev Mal Respir (in French). 29 (1): 64–9. doi:10.1016/j.rmr.2010.03.026. PMID 22240222.
- ↑ Maroto JP, Hudes G, Dutcher JP, et al. (May 2011). "Drug-related pneumonitis in patients with advanced renal cell carcinoma treated with temsirolimus". J. Clin. Oncol. 29 (13): 1750–6. doi:10.1200/JCO.2010.29.2235. PMID 21444868.
- ↑ Errasti P, Izquierdo D, Martín P, et al. (October 2010). "Pneumonitis associated with mammalian target of rapamycin inhibitors in renal transplant recipients: a single-center experience". Transplant. Proc. 42 (8): 3053–4. doi:10.1016/j.transproceed.2010.07.066. PMID 20970608.
- ↑ http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=3275b824-3f82-4151-2ab2-0036a9ba0acc
- ↑ McAlister VC, Mahalati K, Peltekian KM, Fraser A, MacDonald AS (June 2002). "A clinical pharmacokinetic study of tacrolimus and sirolimus combination immunosuppression comparing simultaneous to separated administration.". Ther Drug Monit. 24 (3): 346–50. doi:10.1097/00007691-200206000-00004. PMID 12021624.
- 1 2 3 Schwecke T, Aparicio JF, Molnár I; Aparicio; Molnar; Konig; Khaw; Haycock; Oliynyk; Caffrey; Cortes; Lester; Bohm; Staunton; Leadlay; et al. (August 1995). "The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin". Proc. Natl. Acad. Sci. U.S.A. 92 (17): 7839–43. Bibcode:1995PNAS...92.7839S. doi:10.1073/pnas.92.17.7839. PMC 41241. PMID 7644502.
- ↑ Gregory MA, Gaisser S, Lill RE, et al. (May 2004). "Isolation and characterization of pre-rapamycin, the first macrocyclic intermediate in the biosynthesis of the immunosuppressant rapamycin by S. hygroscopicus". Angew. Chem. Int. Ed. Engl. 43 (19): 2551–3. doi:10.1002/anie.200453764. PMID 15127450.
- ↑ Gregory MA, Hong H, Lill RE, et al. (October 2006). "Rapamycin biosynthesis: Elucidation of gene product function". Org. Biomol. Chem. 4 (19): 3565–8. doi:10.1039/b608813a. PMID 16990929.
- ↑ Graziani EI (May 2009). "Recent advances in the chemistry, biosynthesis and pharmacology of rapamycin analogs". Nat Prod Rep. 26 (5): 602–9. doi:10.1039/b804602f. PMID 19387497.
- ↑ Gatto, G. J., Jr.; Boyne, M. T., II; Kelleher,N. L.; Walsh, C. T. (2006). "Biosynthesis of Pipecolic Acid by RapL, a Lysine Cyclodeaminase Encoded in the Rapamycin Gene Cluster". J. Am. Chem. Soc. 128 (11): 3838–47. doi:10.1021/ja0587603. PMID 16536560.
- ↑ Aparicio JF, Molnár I, Schwecke T, et al. (February 1996). "Organization of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: analysis of the enzymatic domains in the modular polyketide synthase". Gene. 169 (1): 9–16. doi:10.1016/0378-1119(95)00800-4. PMID 8635756.
- ↑ Li, Q; Rao, R; Vazzana, J; Goedegebuure, P; Odunsi, K; Gillanders, W; Shrikant, PA (2012). "Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity". Journal of immunology (Baltimore, Md. : 1950). 188 (7): 3080–7. doi:10.4049/jimmunol.1103365. PMC 3311730. PMID 22379028.
- ↑ Easton, JB; Houghton, PJ (2006). "MTOR and cancer therapy". Oncogene. 25 (48): 6436–46. doi:10.1038/sj.onc.1209886. PMID 17041628.
- ↑ Law BK (October 2005). "Rapamycin: an anti-cancer immunosuppressant?". Crit. Rev. Oncol. Hematol. 56 (1): 47–60. doi:10.1016/j.critrevonc.2004.09.009. PMID 16039868.
- ↑ Fatal AEs Higher with mTOR Drugs in Cancer. Med Page Today
- ↑ Sun SY, Rosenberg LM, Wang X, et al. (August 2005). "Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition". Cancer Res. 65 (16): 7052–8. doi:10.1158/0008-5472.CAN-05-0917. PMID 16103051.
- ↑ Chan S (2004). "Targeting the mammalian target of rapamycin (mTOR): a new approach to treating cancer". Br J Cancer. 91 (8): 1420–4. doi:10.1038/sj.bjc.6602162. PMC 2409926. PMID 15365568.
- ↑ Wendel HG, De Stanchina E, Fridman JS; Stanchina; Fridman; Malina; Ray; Kogan; Cordon-Cardo; Pelletier; Lowe; et al. (March 2004). "Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy". Nature. 428 (6980): 332–7. Bibcode:2004Natur.428..332W. doi:10.1038/nature02369. PMID 15029198. Lay summary – ScienceDaily (18 March 2004).
- ↑ Novak, Kristine (May 2004). "Therapeutics: Means to an end". Nature Reviews Cancer. 4 (5): 332. doi:10.1038/nrc1349.
- ↑ Tuberous Sclerosis Alliance (October 2009). "Current Clinical Trials". Retrieved 14 October 2009.
- ↑ Balestri, R.; Neri, I.; Patrizi, A.; Angileri, L.; Ricci, L.; Magnano, M. (2015-01-01). "Analysis of current data on the use of topical rapamycin in the treatment of facial angiofibromas in Tuberous Sclerosis Complex". Journal of the European Academy of Dermatology and Venereology. 29 (1): 14–20. doi:10.1111/jdv.12665. ISSN 1468-3083.
- ↑ Powers RW, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S (2006). "Extension of chronological life span in yeast by decreased TOR pathway signaling". Genes Dev. 20 (2): 174–84. doi:10.1101/gad.1381406. PMC 1356109. PMID 16418483.
- ↑ Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009). "Rapamycin fed late in life extends lifespan in genetically heterogeneous mice". Nature. 460 (7253): 392–5. doi:10.1038/nature08221. PMC 2786175. PMID 19587680. Lay summary – The Times (8 July 2009).
- ↑ Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, Nadon NL, Strong R (February 2011). "Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice". J. Gerontol. A Biol. Sci. Med. Sci. 66 (2): 191–201. doi:10.1093/gerona/glq178. PMC 3021372. PMID 20974732.
- ↑ Ingram DK, Roth GS (March 2011). "Glycolytic inhibition as a strategy for developing calorie restriction mimetics". Experimental Gerontology (review). 46 (2–3): 148–154. doi:10.1016/j.exger.2010.12.001. PMID 21167272.
- ↑ Tardif, S; Ross, C; Bergman, P; Fernandez, E; Javors, M; Salmon, A; Spross, J; Strong, R; Richardson, A (19 July 2014). "Testing Efficacy of Administration of the Antiaging Drug Rapamycin in a Nonhuman Primate, the Common Marmoset". J Gerontol A Biol Sci Med Sci. 70: 577–588. doi:10.1093/gerona/glu101. PMID 25038772.
- ↑ Check Hayden, Erika (30 October 2014). "Pet dogs set to test anti-ageing drug". Nature. 514 (7524): 546. doi:10.1038/514546a. PMID 25355339. Retrieved 2 April 2015.
- ↑ Amy Harmon (16 May 2016). "Dogs Test Drug Aimed at Humans' Biggest Killer: Age". The New York Times. Retrieved 18 May 2016.
- ↑ Jocelyn Rice (8 July 2009). "First Drug Shown to Extend Life Span in Mammals". Technology Review. Massachusetts Institute of Technology: 1–2. Retrieved 9 July 2009.
- ↑ Lamming, DW (2014). "Diminished mTOR signaling: a common mode of action for endocrine longevity factors". SpringerPlus. 3: 735. doi:10.1186/2193-1801-3-735.
- ↑ Ananieva EA, Powell JD, Hutson SM (2016). "Leucine Metabolism in T Cell Activation: mTOR Signaling and Beyond". Advances in Nutrition. 7 (4): 7985–8055. doi:10.3945/an.115.011221. PMC 4845101. PMID 27422517.
- ↑ Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ (2006). "Hypothalamic mTOR signaling regulates food intake". Science. 312 (5775): 927–930. doi:10.1126/science.1124147. PMID 16690869.
- 1 2 Kriete A, Bosl WJ, Booker G (June 2010). "Rule-Based Cell Systems Model of Aging using Feedback Loop Motifs Mediated by Stress Responses". PLoS Computational Biology. 6 (6): e1000820. doi:10.1371/journal.pcbi.1000820. PMC 2887462. PMID 20585546.
- ↑ Magnuson, Brian; Ekim, Bilgen; Fingar, Diane C. (1 January 2012). "Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks". The Biochemical Journal. 441 (1): 1–21. doi:10.1042/BJ20110892. ISSN 1470-8728. PMID 22168436.
- 1 2 Schieke SM, Phillips D, McCoy JP, Aponte AM, Shen RF, Balaban RS, Finkel T (2006). "The Mammalian Target of Rapamycin (mTOR) Pathway Regulates Mitochondrial Oxygen Consumption and Oxidative Capacity". J. Biol. Chem. 281 (37): 27643–27652. doi:10.1074/jbc.M603536200. PMID 16847060.
- ↑ Ehninger D, Han S, Shilyansky C, et al. "Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis". Nat. Med. 14 (8): 843–8. doi:10.1038/nm1788. PMC 2664098. PMID 18568033.
- ↑ ScienceDaily.com Report
- ↑ Spilman P, Podlutskaya N, Hart MJ, et al. (2010). Ferrari PF, ed. "Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease". PLoS ONE. 5 (4): e9979. doi:10.1371/journal.pone.0009979. PMC 2848616. PMID 20376313.
- ↑ "Nanoparticles treat muscular dystrophy in mice | Newsroom | Washington University in St. Louis". News.wustl.edu. Retrieved 7 April 2014.
- ↑ Fernandez, D; Bonilla, E; Mirza, N; Niland, B; Perl, A (2006). "Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus". Arthritis & Rheumatism. 54 (9): 2983–8. doi:10.1002/art.22085. PMC 4034146. PMID 16947529.
- ↑ Fernandez, D. R.; Telarico, T; Bonilla, E; Li, Q; Banerjee, S; Middleton, F. A.; Phillips, P. E.; Crow, M. K.; Oess, S; Muller-Esterl, W; Perl, A (2009). "Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation". The Journal of Immunology. 182 (4): 2063–73. doi:10.4049/jimmunol.0803600. PMC 2676112. PMID 19201859.
- ↑ Fernandez, D; Perl, A (2010). "MTOR signaling: A central pathway to pathogenesis in systemic lupus erythematosus?". Discovery medicine. 9 (46): 173–8. PMC 3131182. PMID 20350481.
- ↑ Lai, Z. W.; Borsuk, R; Shadakshari, A; Yu, J; Dawood, M; Garcia, R; Francis, L; Tily, H; Bartos, A; Faraone, S. V.; Phillips, P; Perl, A (2013). "Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus". The Journal of Immunology. 191 (5): 2236–46. doi:10.4049/jimmunol.1301005. PMC 3777662. PMID 23913957.
- ↑ Kato, H; Perl, A (2014). "Mechanistic Target of Rapamycin Complex 1 Expands Th17 and IL-4+ CD4-CD8- Double-Negative T Cells and Contracts Regulatory T Cells in Systemic Lupus Erythematosus". The Journal of Immunology. 192 (9): 4134–44. doi:10.4049/jimmunol.1301859. PMC 3995867. PMID 24683191.
- ↑ Caza, T. N.; Fernandez, D. R.; Talaber, G; Oaks, Z; Haas, M; Madaio, M. P.; Lai, Z. W.; Miklossy, G; Singh, R. R.; Chudakov, D. M.; Malorni, W; Middleton, F; Banki, K; Perl, A (2013). "HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE". Annals of the Rheumatic Diseases. 73: 1888–1897. doi:10.1136/annrheumdis-2013-203794. PMID 23897774.
- ↑ Donia M, McCubrey JA, Bendtzen K, Nicoletti F (December 2010). "Potential use of rapamycin in HIV infection". Br J Clin Pharmacol. 70 (6): 784–93. doi:10.1111/j.1365-2125.2010.03735.x. PMC 3014061. PMID 21175433.
- ↑ Campbell GR, Spector SA (March 2011). "Hormonally active vitamin D3 (1{alpha},25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection". J Biol Chem. 286 (21): 18890–902. doi:10.1074/jbc.M110.206110. PMID 21454634.
- ↑ Peces R, Peces C, Pérez-Dueñas V, et al. (16 January 2009). "Rapamycin reduces kidney volume and delays the loss of renal function in a patient with autosomal-dominant polycystic kidney disease". NDT Plus. Oxford Journals. 2 (2): 133–5. doi:10.1093/ndtplus/sfn210. ISSN 1753-0792.
- ↑ CNN.com Clue to kids' early aging disease found, 1 July 2011
- ↑ Rivera, VM; Clackson, T; Natesan, S; Pollock, R; Amara, JF; Keenan, T; Magari, SR; Phillips, T; Courage, NL; Cerasoli F, Jr; Holt, DA; Gilman, M (September 1996). "A humanized system for pharmacologic control of gene expression". Nature Medicine. 2 (9): 1028–32. doi:10.1038/nm0996-1028. PMID 8782462.
External links
| Wikimedia Commons has media related to Sirolimus. |