Point accepted mutation
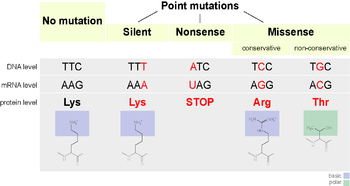
A point accepted mutation — also known as a PAM — is the replacement of a single amino acid in the primary structure of a protein with another single amino acid, which is accepted by the processes of natural selection. This definition does not include all point mutations in the DNA of an organism. In particular, silent mutations are not point accepted mutations, nor are mutations which are lethal or which are rejected by natural selection in other ways.
A PAM matrix is a matrix where each column and row represents one of the twenty standard amino acids. In bioinformatics, PAM matrices are regularly used as substitution matrices to score sequence alignments for proteins. Each entry in a PAM matrix indicates the likelihood of the amino acid of that row being replaced with the amino acid of that column through a series of one or more point accepted mutations during a specified evolutionary interval, rather than these two amino acids being aligned due to chance. Different PAM matrices correspond to different lengths of time in the evolution of the protein sequence.
Biological background
The genetic instructions of every replicating cell in a living organism are contained within its DNA.[1] Throughout the cell's lifetime, this information is transcribed and replicated by cellular mechanisms to produce proteins or to provide instructions for daughter cells during cell division, and the possibility exists that the DNA may be altered during these processes.[1][2] This is known as a mutation. At the molecular level, there are regulatory systems that correct most — but not all — of these changes to the DNA before it is replicated.[2][3]
One of the possible mutations that occurs is the replacement of a single nucleotide, known as a point mutation. If a point mutation occurs within an expressed region of a gene, an exon, then this will change the codon specifying a particular amino acid in the protein produced by that gene.[2] Despite the redundancy in the genetic code, there is a possibility that this mutation will then change the amino acid that is produced during translation, and as a consequence the structure of the protein will be changed.
The functionality of a protein is highly dependent on its structure.[4] Changing a single amino acid in a protein may reduce its ability to carry out this function, or the mutation may even change the function that the protein carries out.[2] Changes like these may severely impact a crucial function in a cell, potentially causing the cell — and in extreme cases, the organism — to die.[5] Conversely, the change may allow the cell to continue functioning albeit differently, and the mutation can be passed on to the organism's offspring. If this change does not result in any significant physical disadvantage to the offspring, the possibility exists that this mutation will persist within the population. The possibility also exists that the change in function becomes advantageous. In either case, while being subjected to the processes of natural selection, the point mutation has been accepted into the genetic pool.
The 20 amino acids translated by the genetic code vary greatly by the physical and chemical properties of their side chains.[4] However, these amino acids can be categorised into groups with similar physicochemical properties.[4] Substituting an amino acid with another from the same category is more likely to have a smaller impact on the structure and function of a protein than replacement with an amino acid from a different category. Consequently, acceptance of point mutations depends heavily on the amino acid being replaced in the mutation, and the replacement amino acid. The PAM matrices are a mathematical tool that account for these varying rates of acceptance when evaluating the similarity of proteins during alignment.
Terminology
The term accepted point mutation was initially used to describe the mutation phenomenon. However, the acronym PAM was preferred over APM due to readability, and so the term point accepted mutation is used more regularly.[6] Because the value in the PAMn matrix represents the number of mutations per 100 amino acids, which can be likened to a percentage of mutations, the term percentage accepted mutation is sometimes used.

It is important to distinguish between point accepted mutations (PAMs), point accepted mutation matrices (PAM matrices) and the PAMn matrix. The term 'point accepted mutation' refers to the mutation event itself. However, 'PAM matrix' refers to one of a family of matrices which contain scores representing the likelihood of two amino acids being aligned due to a series of mutation events, rather than due to random chance. The 'PAMn matrix' is the PAM matrix corresponding to a time frame long enough for mutation events to occur per 100 amino acids.
Construction of PAM matrices
PAM matrices were introduced by Margaret Dayhoff in 1978.[7] The calculation of these matrices were based on 1572 observed mutations in the phylogenetic trees of 71 families of closely related proteins. The proteins to be studied were selected on the basis of having high similarity with their predecessors. The protein alignments included were required to display at least 85% identity.[6][8] As a result, it is reasonable to assume that any aligned mismatches were the result of a single mutation event, rather than several at the same location.
Each PAM matrix has twenty rows and twenty columns — one representing each of the twenty amino acids translated by the genetic code. The value in each cell of a PAM matrix is related to the probability of a column amino acid before the mutation being aligned with a row amino acid afterwards.[6][7][8] From this definition, PAM matrices are an example of a substitution matrix.
Collection of data from phylogenetic trees
For each branch in the phylogenetic trees of the protein families, the number of mismatches that were observed were recorded and a record kept of the two amino acids involved.[7] These counts were used as entries below the main diagonal of the matrix . Since the vast majority of protein samples come from organisms that are alive today (extant species), the 'direction' of a mutation cannot be determined. That is, the amino acid present before the mutation cannot be distinguished from the amino acid that replaced it after the mutation. Because of this, the matrix is assumed to be symmetric, and the entries of above the main diagonal are computed on this basis. The entries along the diagonal of do not correspond to mutations and can be left unfilled.

In addition to these counts, data on the mutability and the frequency of the amino acids was obtained.[6][7] The mutability of an amino acid is the ratio of the number of mutations it is involved in and the number of times it occurs in an alignment.[7] Mutability measures how likely an amino acid is to mutate acceptably. Asparagine, an amino acid with a small polar side chain, was found to be the most mutable of the amino acids.[7] Cysteine and tryptophan were found to be the least mutable amino acids.[7] The side chains for cysteine and tryptophan have less common structures: cysteine's side chain contains sulfur which participates in disulfide bonds with other cysteine molecules, and tryptophan's side chain is large and aromatic.[4] Since there are several small polar amino acids, these extremes suggest that amino acids are more likely to acceptably mutate if their physical and chemical properties are more common among alternative amino acids.[6][8]
Construction of the mutation matrix
For the th amino acid, the values and are its mutability and frequency. The frequencies of the amino acids are normalised so that they sum to 1. If total number of occurrences of the th amino acid is , and is the total number of all amino acids, then






Based on the definition of mutability as the ratio of mutations to occurrences of an amino acid

or

The mutation matrix is constructed so that the entry represents the probability of the th amino acid mutating into the th amino acid. The non-diagonal entries are computed by the equation[7]




where is a constant of proportionality. However, this equation does not compute the diagonal entries. Each column in the matrix lists each of the twenty possible outcomes for an amino acid — it can mutate into one of the 19 other amino acids, or remain unchanged. Since the non-diagonal entries listing the probabilities of each of the 19 mutations are known, and the sum of the probabilities of these twenty outcomes must be 1, this last probability can be calculated by


which simplifies to[7]

Calculation of the diagonal entries Substituting in the expression for the non-diagonal entries mutation matrix:
Since the values of and are constants that don't change with the value of
And thus cancellation reveals that



A result of particular significance is that for the non-diagonal entries

Which means that for all entries in the mutation matrix

Choice of the constant of proportionality
The probabilities contained in vary as some unknown function of the amount of time that a protein sequence is allowed to mutate for. Instead of attempting to determine this relationship, the values of are calculated for a short time frame, and the matrices for longer periods of time are calculated by assuming mutations follow a Markov chain model.[9][10] The base unit of time for the PAM matrices is the time required for 1 mutation to occur per 100 amino acids, sometimes called 'a PAM unit' or 'a PAM' of time.[6] This is precisely the duration of mutation assumed by the PAM1 matrix.
The constant is used to control the proportion of amino acids that are unchanged. By using only alignments of proteins that had at least 85% similarity, it could be reasonably assumed that the mutations observed were direct, without any intermediate states. This means that scaling down these counts by a common factor would provide an accurate estimate of the mutation counts had the similarity been closer to 100%. It also means that the number of mutations per 100 amino acids, the in PAMn is equal to the number of mutated amino acids per 100 amino acids.
To find the mutation matrix for the PAM1 matrix, the requirement that 99% of the amino acids in a sequence are conserved is imposed. The quantity is equal to the number of conserved amino acid units, and so the total number of conserved amino acids is


The value of needed to be pick to produce 99% identity after mutation is then given by the equation

This value can then be used in the mutation matrix for the PAM1 matrix.
Construction of the PAMn matrices
The Markov chain model of protein mutation relates the mutation matrix for PAMn, , to the mutation matrix for the PAM1 matrix, by the simple relationship



The PAMn matrix is constructed from the ratio of the probability of point accepted mutations replacing the th amino acid with the th amino acid, to the probability of these amino acids being aligned by chance. The entries of the PAMn matrix are given by the equation[11][12]

Note that in Gusfield's book, the entries and are related to the probability of the th amino acid mutating into the th amino acid.[11] This is the origin of the different equation for the entries of the PAM matrices.

When using the PAMn matrix to score an alignment of two proteins, the following assumption is made:
- If these two proteins are related, the evolutionary interval separating them is the time taken for point accepted mutations to occur per 100 amino acids.
When the alignment of the th and th amino acids is considered, the score indicates the relative likelihoods of the alignment due to the proteins being related or due to random chance.
- If the proteins are related, a series of point accepted mutations must have occurred to mutate the original amino acid into its replacement. Suppose the th amino acid is the original. Based on the abundance of amino acids in proteins, the probability of the th amino acid being the original is . Given any particular unit of this amino acid, the probability of being replaced by the th amino acid in the assumed time interval is . Thus, the probability of the alignment is , the numerator within the logarithm.
- If the proteins are not related, the events that the two aligned amino acids are the th and th amino acids must be independent.The probabilities of these events are and , which means the probability of the alignment is , the denominator of the logarithm.
- Thus, the logarithm in the equation results in a positive entry if the alignment is more likely due to point accepted mutations, and a negative entry if the alignment is more likely due to chance.




Properties of the PAM matrices
Symmetry of the PAM matrices
While the mutation probability matrix is not symmetric, each of the PAM matrices are.[6][7] This somewhat surprising property is a result of the relationship that was noted for the mutation probability matrix:

In fact, this relationship holds for all positive integer powers of the matrix :
Generalisation of property to positive integer matrix powers This generalisation can be proven using mathematical induction. Suppose that for a matrix
And that for a positive integer
By expansion of the matrix product ,
Using the property we have assumed of the matrix
And using the property for the matrix
In this case, it is only known at first that the result holds for . However, the above argument shows that the property also holds for . This new knowledge then shows that the property also holds for and this repeats to show that the property holds for all positive integers .














As a result, the entries of the PAMn matrix are symmetric, since

Relating the number of mutated amino acids and the number of mutations
The value represents the number of mutations that occur per 100 amino acids, however this value is rarely accessible and often estimated. However, when comparing two proteins it is easy to calculate instead, which is the number of mutated amino acids per 100 amino acids. Despite the random nature of mutation, these values can be approximately related by[13]


Derivation of relationship between and Mutations in the primary structure of a protein can occur anywhere along the sequence. If it is assumed the distribution of the mutations among amino acid positions is uniform, the problem is analogous to a distribution of "balls into bins", a common problem in combinatorics. In a case where balls (i.e. mutations) are distributed amongst bins (amino acid positions), the number of bins containing at least one ball, has a distribution with a mean given by[14]
If the rate of mutation is mutations per 100 amino acids, then
And if there are mutated amino acids per 100 amino acids, then it is approximately equal to
Now and can be related by
For large values of , an assumption that can be reasonably made for typical proteins, this expression is approximately equal to






The validity of these estimates can be verified by counting the number of amino acids that remain unchanged under the action of the matrix . The total number of unchanged amino acids for the time interval of the PAMn matrix is

and so the proportion of unchanged amino acids is

An example - PAM250
A PAM250 is a commonly used scoring matrix for sequence comparison. Only the lower half of the matrix needs to be computed, since by their construction, PAM matrices are required to be symmetric. Each of the 20 amino acid are shown down the top and side of the matrix, with 3 additional ambiguous amino acids. The amino acids are most commonly shown listed alphabetically, or listed in groups. These groups are the characteristics shared among the amino acids.[7]
Uses in bioinformatics
Determining the time of divergence in phylogenetic trees
The molecular clock hypothesis predicts that the rate of amino acid substitution in a particular protein will be approximately constant over time, though this rate may vary between protein families.[13] This suggests that the number of mutations per amino acid in a protein increases approximately linearly with time.
Determining the time at which two proteins diverged is an important task in phylogenetics. Fossil records are often used to establish the position of events on the timeline of the Earth's evolutionary history, but the application of this source is limited. However, if the rate at which the molecular clock of protein family ticks — that is, the rate at which the number of mutations per amino acid increases — is known, then knowing this number of mutations would allow the date of divergence to be found.
Suppose the date of divergence for two related proteins, taken from organisms living today, is sought. The two proteins have both been accumulating accepted mutations since the date of divergence, and so the total number of mutations per amino acid separating them is approximately twice that which separates them from their common ancestor. If a range of PAM matrices are used to align two proteins that are known to be related, then the value of in the PAMn matrix which results in the best score is most likely to correspond to the mutations per amino acid separating the two proteins. Halving this value and dividing by the rate at which accepted mutations accumulate in the protein family provides an estimate of the time of divergence of these two proteins from their common ancestor. That is, the time of divergence in myr is[13]

Where is the number of mutations per amino acid, and is the rate of accepted mutation accumulation in mutations per amino acid site per million years.

Use in BLAST
PAM matrices are also used as a scoring matrix when comparing DNA sequences or protein sequences to judge the quality of the alignment. This form of scoring system is utilized by a wide range of alignment software including BLAST.[15]
Comparing PAM and BLOSUM
In addition to PAM matrices, another more recently developed scoring matrix can be used. This is known as a BLOSUM. The two result in the same scoring outcome, but use differing methodologies. BLOSUM directly look at mutations in motifs of related sequences while PAM's extrapolate evolutionary information based on closely related sequences.[16]
Since both PAM and BLOSUM are different methods for showing the same scoring information, the two can be compared but due to the very different method of obtaining this score, a PAM100 does not equal a BLOSUM100.[17]
| PAM | BLOSUM |
|---|---|
| PAM100 | BLOSUM90 |
| PAM120 | BLOSUM80 |
| PAM160 | BLOSUM60 |
| PAM200 | BLOSUM52 |
| PAM250 | BLOSUM45 |
See also
References
- 1 2 Campbell NA, Reece JB, Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). "The Molecular Basis of Inheritance". Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 307–325. ISBN 9781442502215.
- 1 2 3 4 Campbell NA, Reece JB, Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). "From Gene to Protein". Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 327–350. ISBN 9781442502215.
- ↑ Pal JK, Ghaskadbi SS (2009). "DNA Damage, Repair and Recombination". Fundamentals of Molecular Biology (1st ed.). Oxford University Press. pp. 187–203. ISBN 9780195697810.
- 1 2 3 4 Campbell NA, Reece JB, Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). "The Structure and Function of Large Biological Molecules". Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 68–89. ISBN 9781442502215.
- ↑ Lobo, Ingrid (2008). "Mendelian Ratios and Lethal Genes". Nature. Nature Publishing Group. Retrieved 19 October 2013.
- 1 2 3 4 5 6 7 Pevsner J (2009). "Pairwise Sequence Alignment". Bioinformatics and Functional Genomics (2nd ed.). Wiley-Blackwell. pp. 58–68. ISBN 978-0-470-08585-1.
- 1 2 3 4 5 6 7 8 9 10 11 Dayhoff, M.O., Schwartz, R. and Orcutt, B.C. (1978). "A model of Evolutionary Change in Proteins". Atlas of protein sequence and structure (volume 5, supplement 3 ed.). Nat. Biomed. Res. Found. pp. 345–358. ISBN 0-912466-07-3.
- 1 2 3 Wing-Kin Sung (2010). Algorithms in Bioinformatics: A Practical Introduction. CRC Press. pp. 51–52. ISBN 978-1-4200-7033-0.
- ↑ Kosiol C, Goldman N (2005). "Different versions of the Dayhoff rate matrix.". Molecular biology and evolution. 22 (2): 193–9. doi:10.1093/molbev/msi005. PMID 15483331.
- ↑ Liò P, Goldman N (1998). "Models of molecular evolution and phylogeny". Genome Research. 8 (12): 1233–44. doi:10.1101/gr.8.12.1233. PMID 9872979.
- 1 2 Gusfield, Dan (1997). Algorithms on String, Trees, and Sequences -Computer Science and Computational Biology. Cambridge University Press. pp. 383–384. ISBN 0521585198.
- ↑ Boeckenhauer, Hans-Joachim; Dirk Bongartz (2010). Algorithmic Aspects of Bioinformatics. Springer. pp. 94–96. ISBN 3642091008.
- 1 2 3 Pevsner J (2009). "Molecular Phylogeny and Evolution". Bioinformatics and Functional Genomics (2nd ed.). Wiley-Blackwell. pp. 221–227. ISBN 978-0-470-08585-1.
- ↑ Motwani, Rajeev; Prabhakar Raghavan (1995). Randomized Algorithms. Cambridge University Press. p. 94. ISBN 0521474655.
- ↑ "The Statistics of Sequence Similarity Scores". National Centre for Biotechnology Information. Retrieved 20 October 2013.
- ↑ Henikoff S, Henikoff JG (1992). "Amino acid substitution matrices from protein blocks." (PDF). Proceedings of the National Academy of Sciences of the United States of America. 89 (22): 10915–10919. doi:10.1073/pnas.89.22.10915. PMC 50453
 . PMID 1438297.
. PMID 1438297. - ↑ Saud, Omama (2009). "PAM and BLOSSUM SUBSITUTION MATRICES". Birec. Retrieved 20 October 2013.
External links
- http://www.inf.ethz.ch/personal/gonnet/DarwinManual/node148.html
- http://www.bioinformatics.nl/tools/pam.html For quickly calculating a PAM matrix.
- http://web.expasy.org/docs/relnotes/relstat.html The most recent statistics from the Swiss-Prot protein knowledgebase. Section 6.1 contains the most up-to-date amino acid frequencies