Transition metal oxo complex
In coordination chemistry, an oxo ligand is an oxygen atom bound only to one or more metal centers. These ligands can exist as terminal or (most commonly) as bridging atom (Fig. 1). Oxo ligands stabilize high oxidation states of a metal.[1]
Oxo ligands are pervasive, comprising the great majority of the Earth's crust. This article concerns a subset of oxides, molecular derivatives. They are also found in several metalloenzymes, e.g. in the molybdenum cofactor and in many iron-containing enzymes. One of the earliest synthetic compounds to incorporate an oxo ligand is sodium ferrate (Na2FeO4) circa 1702.[2]
Reactivity
Olation and acid-base reactions
Common reactions affected by metal-oxo compounds is olation, the condensation process that converts low molecular weight oxides to polymeric materials, including minerals. Olation often begins with the deprotonation of a metal-hydroxo complex.
Oxygen-atom transfer
Oxygen-atom transfer is common reaction of particular interest in organic chemistry and biochemistry.[3] Some metal-oxos are capable of transferring their oxo ligand to organic substrates. One such example of this type of reactivity is from and enzyme super-family Molybdenum oxotransferase.
Hydrogen Atom Abstraction
Transition metal-oxo's are also capable of abstracting strong C–H, N–H, and O–H bonds. Cytochrome P450 contains a high-valent iron-oxo which is capable of abstracting hydrogen atoms from strong C–H bonds.[4]
Molecular oxides
Some of the longest known and most widely used oxo compounds are oxidizing agents such as potassium permanganate (KMnO4) and osmium tetroxide (OsO4).[5] Compounds such as these are widely used for converting alkenes to vicinal diols and alcohols to ketones or carboxylic acids.[1] More selective or gentler oxidizing reagents include pyridinium chlorochromate (PCC) and pyridinium dichromate (PDC).[1] Metal oxo species are capable of catalytic, including asymmetric oxidations of various types. Some metal-oxo complexes promote C-H bond activation, converting hydrocarbons to alcohols.[6]
 Selection of molecular metal oxides. From left, vanadyl chloride (d0), a tungsten oxo carbonyl (d2), permanganate (d0), [ReO2(pyridine)4]+ (d2), simplified view of compound I (a state of cytochrome P450, d4), and trismesityliridium oxide (d4).
Selection of molecular metal oxides. From left, vanadyl chloride (d0), a tungsten oxo carbonyl (d2), permanganate (d0), [ReO2(pyridine)4]+ (d2), simplified view of compound I (a state of cytochrome P450, d4), and trismesityliridium oxide (d4).
Metalloenzymes
Iron(IV)-oxo species

Iron(IV)-oxo compounds are intermediates in many oxidations catalysed by heme-containing enzymes. One of the most widely studied examples is cytochrome p450 enzymes, which use a heme cofactor that is capable of hydroxylation of saturated C–H bonds,[7] epoxidation of olefins,[8][9] and oxidation of aromatic groups.[10] Similarly, methane monooxygenase (MMO) oxidizes methane to methanol via oxygen atom transfer from an iron-oxo intermediate at its non-heme di-iron center. First, C-H bonds are quite resistant to oxidation and are generally unreactive at moderate temperatures (see C-H bond activation). Second, harsh oxidizing agents will generally oxidize an alcohol to a carboxylic acid, but these enzymes are able to oxidize an alkyl group to an alcohol without further oxidation to a carbonyl or carboxylic acid. The oxidant used in these enzymatic reactions is molecular oxygen in contrast with the harsh, toxic chemicals often found in conventional synthetic organic oxidations.[11] As is generally the case with enzymatic reactions, these oxidations are chemically selective and take place at fast rates in aqueous solvent. Much of the effort in producing synthetic C-H bond activation catalysts has been inspired by these well designed natural catalysts.[6]
Molybdenum/tungsten oxo species
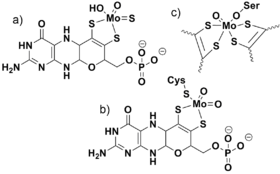
The oxo ligand (or analogous sulfido ligand) is nearly ubiquitous in molybdenum and tungsten chemistry, appearing in the ores containing these elements, throughout their synthetic chemistry, and also in their biological role (aside from nitrogenase). The biologically transported species and starting point for biosynthesis is generally accepted to be oxometallates MoO4−2 or WO4−2. All Mo/W enzymes, again except nitrogenase, are bound to one or more molybdopterin prosthetic group. The Mo/W centers generally cycle between hexavalent (M(IV)) and tetravalent (M(VI)) states. Although there is some variation among these enzymes, members from all three families involve oxygen atom transfer between the Mo/W center and the substrate.[12] Representative reactions from each of the three structural classes are:
- Sulfite oxidase: SO3−2 + H2O → SO4−2 + 2 H+ + 2 e−
- DMSO reductase: H3CS(O)CH3 (DMSO) + 2 H+ + 2 e− → H3CSCH3 (DMS) + H2O
- Aldehyde ferredoxin oxidoreductase: RCHO + H2O → RCO2H + 2 H+ + 2 e−
The three different classes of molybdenum cofactors are shown in the Figure. The biological use of tungsten mirrors that of molybdenum.[13]
Oxygen-evolving complex
The active site for the oxygen-evolving complex (OEC) of photosystem II (PSII) is a Mn4O5Ca centre with several bridging oxo ligands that participate in the oxidation of water to molecular oxygen.[14] The OEC is proposed to utilize a terminal oxo intermediate as a part of the water oxidation reaction.[15] This complex is responsible for the production of nearly all of earth's molecular oxygen. This key link in the oxygen cycle is necessary for much of the biodiversity present on earth.

The "oxo wall"
.png)
The term "oxo wall" is a theory used to describe the fact that no terminal oxo complexes are known for metal centers with tetragonal symmetry and d-electron counts beyond 5.[17] Oxo compounds for the vanadium through iron triads (groups 3-8) are well known, whereas terminal oxo compounds for metals in the cobalt through zinc triads (groups 9-12) are rare and invariably feature metals with coordination numbers lower than 6. This trend holds for other metal-ligand multiple bonds. Claimed exceptions to this rule have been retracted.[18]
Terminal oxo ligands are also rather rare for the titanium triad, especially zirconium and hafnium and is unknown for group 3 metals (scandium, yttrium, and lanthanum).[1]
At first glance, the iridium oxo complex Ir(O)(mesityl)3 may appear to be an exception to the oxo-wall, but it is not.[19] The complex has trigonal symmetry, which produces a reordering of the metal d-orbitals below the degenerate MO pi* pair. In three-fold symmetric complexes, multiple MO bonding is allowed for as many as 7 d-electrons.[17]
See also
References
- 1 2 3 4 Nugent, W. A., Mayer, J. M. "Metal-Ligand Multiple Bonds." John Wiley & Sons, New York, 1988.
- ↑ Sharpless, K.B.; Flood, T.C. (1971). "Oxotransition metal oxidants as mimics for the action of mixed-function oxygenases. 'NIH shift' with chromyl reagents". J. Am. Chem. Soc. 93 (9): 2316–8. doi:10.1021/ja00738a039. PMID 5553075.
- ↑ Holm, R. H. (1987). "Metal-centered oxygen atom transfer reactions". Chem. Rev. 87 (6): 1401–1449. doi:10.1021/cr00082a005.
- ↑ Meunier, Bernard; de Visser, Samuël P.; Shaik, Sason (2004). "Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes". Chemical Reviews. 104 (9): 3947–3980. doi:10.1021/cr020443g. ISSN 0009-2665. PMID 15352783.
- ↑ Du, G. & Abu-Omar, M.M. (2008). "Oxo and Imido Complexes of Rhenium and Molybdenum in Catalytic Reductions". Current Organic Chemistry. 12 (14): 1185–1198. doi:10.2174/138527208785740238.
- 1 2 Gunay A. & Theopold, K.H. (2010). "C-H Bond Activations by Metal Oxo Compounds". Chem. Rev. 110 (2): 1060–1081. doi:10.1021/cr900269x.
- ↑ Ortiz de Montellano, Paul R. (2010). "Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes". Chemical Reviews. 110 (2): 932–948. doi:10.1021/cr9002193. ISSN 0009-2665. PMC 2820140
 . PMID 19769330.
. PMID 19769330. - ↑ Coon, M. J. (1998-01-20). "Epoxidation of olefins by cytochrome P450: Evidence from site-specific mutagenesis for hydroperoxo-iron as an electrophilic oxidant". Proceedings of the National Academy of Sciences. 95: 3555–60. doi:10.1073/pnas.95.7.3555. PMID 9520404.
- ↑ Farinas, Edgardo T; Alcalde, Miguel; Arnold, Frances (2004). "Alkene epoxidation catalyzed by cytochrome P450 BM-3 139-3". Tetrahedron. 60 (3): 525–528. doi:10.1016/j.tet.2003.10.099. ISSN 0040-4020.
- ↑ Korzekwa, Kenneth; Trager, William; Gouterman, Martin; Spangler, Dale; Loew, Gilda (1985). "Cytochrome P450 mediated aromatic oxidation: a theoretical study". Journal of the American Chemical Society. 107 (14): 4273–4279. doi:10.1021/ja00300a033. ISSN 0002-7863.
- ↑ Brunold, T.C. (2007). "Synthetic iron-oxo 'diamond core' mimics structure of key intermediate in methane monooxygenase catalytic cycle". Proc. Natl. Acad. Sci. U.S.A. 104 (52): 20641–20642. Bibcode:2007PNAS..10420641B. doi:10.1073/pnas.0710734105.
- ↑ Schwarz, G., Mendel, R.R., and Ribbe, M.W. (2009). "Molybdenum cofactors, enzymes and pathways". Nature. 460 (7257): 839–847. Bibcode:2009Natur.460..839S. doi:10.1038/nature08302.
- ↑ Mukund, S. & Adams, M.W.W. (1996). "Molybdenum and Vanadium Do Not Replace Tungsten in the Catalytically Active Forms of the Three Tungstoenzymes in the Hyperthermophilic Archaeon Pyrococcus furiosus". J. Bact.: 163–167.
- ↑ Umena, Yasufumi; Kawakami, Keisuke; Shen, Jian-Ren; Kamiya, Nobuo (2011). "Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å". Nature. 473 (7345): 55–60. doi:10.1038/nature09913. ISSN 0028-0836. PMID 21499260.
- ↑ Umena, Yasufumi; Kawakami, Keisuke; Shen, Jian-Ren; Kamiya, Nobuo (2011). "Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å". Nature. 473: 55–60. doi:10.1038/nature09913. PMID 21499260.
- ↑ Umena, Yasufumi; Kawakami, Keisuke; Shen, Jian-Ren; Kamiya, Nobuo. "Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å". Nature. 473 (7345): 55–60. doi:10.1038/nature09913.
- 1 2 Winkler, J. R.; Gray, H. B. (2012). "Electronic Structures of Oxo-Metal Ions". Struct. Bond. Structure and Bonding. 142: 17–28. doi:10.1007/430_2011_55. ISBN 978-3-642-27369-8.
- ↑ o’Halloran, Kevin P.; Zhao, Chongchao; Ando, Nicole S.; Schultz, Arthur J.; Koetzle, Thomas F.; Piccoli, Paula M. B.; Hedman, Britt; Hodgson, Keith O.; et al. (2012). "Revisiting the Polyoxometalate-Based Late-Transition-Metal-Oxo Complexes: The "Oxo Wall" Stands". Inorganic Chemistry. 51 (13): 7025–31. doi:10.1021/ic2008914. PMID 22694272.
- ↑ Hay-Motherwell, R. S.; Wilkinson, G.; Hussain-Bates, B.; Hursthouse, M. B. (1993). "Synthesis and X-ray Crystal Structure of Oxotrimesityl-Iridium(V)". Polyhedron. 12 (16): 2009–2012. doi:10.1016/S0277-5387(00)81474-6.