Mulibrey nanism
| Mulibrey nanism | |
|---|---|
| Classification and external resources | |
| OMIM | 253250 |
| DiseasesDB | 32892 |
Mulibrey nanism ("MUscle-LIver-BRain-EYe nanism"), also called Perheentupa syndrome and pericardial constriction with growth failure,[1] is a rare autosomal recessive congenital disorder.[2] It causes severe growth failure (dwarfism) along with abnormalities of the heart, muscle, liver, brain and eye.[3] Mulibrey nanism has been associated with a recessive defect in the gene TRIM37.[4] TRIM37 is responsible for various cellular functions including developmental patterning.[5]
Characteristics
A person with Mulibrey nanism has growth retardation, a short broad neck, misshapen sternum, small thorax, square shoulders, triangular face, unusual voice, enlarged liver, and yellowish dots in the ocular fundi.[6] Individuals with Mulibrey nanism have also been reported to have mental retardation, tumors, and infertility.[6]
Cause and genetics
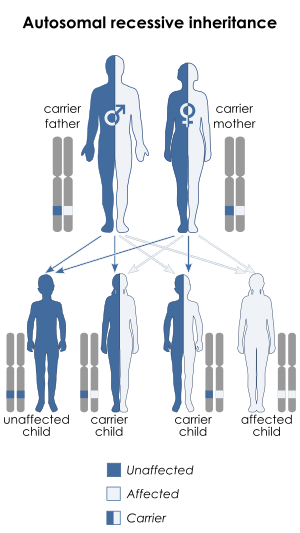
Mulibrey nanism is caused by mutations of the TRIM37 gene, located at human chromosome 17q22-23.[2][7] The disorder is inherited in an autosomal recessive manner.[2] This means the defective gene responsible for the disorder is located on an autosome (chromosome 17 is an autosome), and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Natural history
A rare disease, Mulibrey nanism may be a Finnish heritage disease. Worldwide, it has been documented in 110 persons, 85 of them Finnish. It is a recessive genetic disease. Many people with Mulibrey nanism have parents who are closely related (consanguine). Signs and symptoms are variable. Siblings who suffer this disease sometimes do not share the same symptoms.[6]
Treatment
Drug therapies can relieve some of the symptoms and the pain related to this disease.[8]
References
- ↑ Online Mendelian Inheritance in Man (OMIM) 253250
- 1 2 3 Doganc TM, Y. K. B.; Yüksel Konuk, B.; Alpan, N.; Konuk, O.; Hämäläinen, R.; Lehesjoki, A.; Tekin, M. (July 2007). "A novel mutation in TRIM37 is associated with mulibrey nanism in a Turkish boy". Clinical Dysmorphology. 16 (3): 173–176. doi:10.1097/MCD.0b013e3280f6d00b. PMID 17551331.
- ↑ Hes FJ, Morreau H (Jun 2009). "Where genetics and pathology meet: Mulibrey nanism". The Journal of Pathology. 218 (2): 143–145. doi:10.1002/path.2552. PMID 19347900.
- ↑ Jagiello P, Hammans C, Wieczorek S, et al. (Jun 2003). "A novel splice site mutation in the TRIM37 gene causes mulibrey nanism in a Turkish family with phenotypic heterogeneity". Hum. Mutat. 21 (6): 630–635. doi:10.1002/humu.10220. PMID 12754710.
- ↑ "TRIM37 tripartite motif-containing 37". NCBI Entrez Gene. Retrieved 2009-05-07.
- 1 2 3 OMIM (1966-2009). Mulibrey nanism. NCBI (Johns Hopkins University). Retrieved May 7, 2009 from, http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=253250
- ↑ Online Mendelian Inheritance in Man (OMIM) 605073
- ↑ WD (2009). Treatments for mulibrey nanism syndrome. Health Grades Inc. Retrieved May 7, 2009 from, http://www.wrongdiagnosis.com/m/mulibrey_nanism_syndrome/treatments.htm
External links
- Mulibrey Nanism syndrome at NIH's Office of Rare Diseases
- Mulibrey nanism: clinical features and diagnostic criteria
- Growth and Growth Hormone Therapy in Subjects With Mulibrey Nanism