Mitochondrial disease
| Mitochondrial disease | |
|---|---|
|
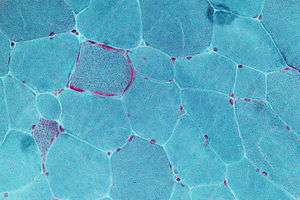
Micrograph showing ragged red fibers, a finding seen in various types of mitochondrial diseases. Muscle biopsy. Gomori trichrome stain. | |
| Classification and external resources | |
| Specialty | Medical genetics |
| ICD-9-CM | 277.87 |
| DiseasesDB | 28840 |
| MeSH | D028361 |
Mitochondrial disease is a group of disorders caused by dysfunctional mitochondria, the organelles that generate energy for the cell. Mitochondria are found in every cell of the human body except red blood cells, and convert the energy of food molecules into the ATP that powers most cell functions.
Mitochondrial diseases are sometimes (about 15% of the time)[1] caused by mutations in the mitochondrial DNA that affect mitochondrial function. Other causes of mitochondrial disease are mutations in genes of the nuclear DNA, whose gene products are imported into the mitochondria (mitochondrial proteins) as well as acquired mitochondrial conditions. Mitochondrial diseases take on unique characteristics both because of the way the diseases are often inherited and because mitochondria are so critical to cell function. The subclass of these diseases that have neuromuscular disease symptoms are often called a mitochondrial myopathy.
Signs and symptoms
Symptoms include poor growth, loss of muscle coordination, muscle weakness, visual problems, hearing problems, learning disabilities, heart disease, liver disease, kidney disease, gastrointestinal disorders, respiratory disorders, neurological problems, autonomic dysfunction and dementia.
The body, and each mutation, is modulated by other genome variants; the mutation that in one individual may cause liver disease might in another person cause a brain disorder. The severity of the specific defect may also be great or small. Some minor defects cause only "exercise intolerance", with no serious illness or disability. Defects often affect the operation of the mitochondria and multiple tissues more severely, leading to multi-system diseases.
As a rule, mitochondrial diseases are worse when the defective mitochondria are present in the muscles, cerebrum, or nerves,[2] because these cells use more energy than most other cells in the body.
Although mitochondrial diseases vary greatly in presentation from person to person, several major clinical categories of these conditions have been defined, based on the most common phenotypic features, symptoms, and signs associated with the particular mutations that tend to cause them.
An outstanding question and area of research is whether ATP depletion or reactive oxygen species are in fact responsible for the observed phenotypic consequences.
Causes
Mitochondrial disorders may be caused by mutations, acquired or inherited, in mitochondrial DNA (mtDNA) or in nuclear genes that code for mitochondrial components. They may also be the result of acquired mitochondrial dysfunction due to adverse effects of drugs, infections, or other environmental causes (see MeSH).
Nuclear DNA has two copies per cell (except for sperm and egg cells), one copy being inherited from the father and the other from the mother. Mitochondrial DNA, however, is strictly inherited from the mother and each mitochondrial organelle typically contains multiple mtDNA copies. During cell division the existing mitochondria segregate randomly between the two new cells, and then those mitochondria make more copies. As mtDNA is copied when mitochondria proliferate, they can accumulate random mutations. If only a few of the mtDNA copies inherited from the mother are defective, mitochondrial division may cause most of the defective copies to end up in just one of the new mitochondria (for more detailed inheritance patterns, see Human mitochondrial genetics). Mitochondrial disease may become clinically apparent once the number of affected mitochondria reaches a certain level; this phenomenon is called "threshold expression".
Mitochondrial DNA mutations occur frequently, due to the lack of the error checking capability that mtDNA has (see Mutation rate). This means that mitochondrial DNA disorders may occur spontaneously and relatively often. Defects in enzymes that control mitochondrial DNA replication (all of which are encoded for by genes in the nuclear DNA) may also cause mitochondrial DNA mutations.
Most mitochondrial function and biogenesis is controlled by nuclear DNA. Human mitochondrial DNA encodes only 13 proteins of the respiratory chain, while most of the estimated 1,500 proteins and components targeted to mitochondria are nuclear-encoded. Defects in nuclear-encoded mitochondrial genes are associated with hundreds of clinical disease phenotypes including anemia, dementia, hypertension, lymphoma, retinopathy, seizures, and neurodevelopmental disorders.[3]
A study by Yale University researchers published in the Feb 12, 2004 issue of the New England Journal of Medicine explores the role of mitochondria in insulin resistance among the offspring of patients with type 2 diabetes.[4] Other studies have shown that the mechanism may involve the interruption of the mitochondrial signaling process in body cells (intramyocellular lipids). A study conducted at the Pennington Biomedical Research Center in Baton Rouge, LA (Diabetes 54, 2005 1926-33) showed that this in turn partially disables the genes that produce mitochondria.
Examples
Examples of mitochondrial diseases include:
- Mitochondrial myopathy
- Diabetes mellitus and deafness (DAD)
- this combination at an early age can be due to mitochondrial disease
- Diabetes mellitus and deafness can also be found together for other reasons
- Leber's hereditary optic neuropathy (LHON)
- visual loss beginning in young adulthood
- eye disorder characterized by progressive loss of central vision due to degeneration of the optic nerves and retina
- affects 1 in 50,000 people in Finland
- Leigh syndrome, subacute sclerosing encephalopathy
- after normal development the disease usually begins late in the first year of life, although onset may occur in adulthood
- a rapid decline in function occurs and is marked by seizures, altered states of consciousness, dementia, ventilatory failure
- Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP)
- progressive symptoms as described in the acronym
- dementia
- Myoneurogenic gastrointestinal encephalopathy (MNGIE)
- gastrointestinal pseudo-obstruction
- neuropathy
- Myoclonic Epilepsy with Ragged Red Fibers (MERRF)
- progressive myoclonic epilepsy
- "Ragged Red Fibers" – clumps of diseased mitochondria accumulate in the subsarcolemmal region of the muscle fiber and appear as "Ragged Red Fibers" when muscle is stained with modified Gömöri trichrome stain
- short stature
- hearing loss
- lactic acidosis
- exercise intolerance
- Mitochondrial myopathy, encephalomyopathy, lactic acidosis, stroke-like symptoms (MELAS)
- mtDNA depletion
Nota bene: Conditions such as Friedreich's ataxia can affect the mitochondria, but are not associated with mitochondrial proteins.
Mechanisms
The effective overall energy unit for the available body energy is referred to as the daily glycogen generation capacity,[5][6][7] and is used to compare the mitochondrial output of healthy individuals to that of afflicted or chronically glycogen-depleted individuals. This value is slow to change in a given individual, as it takes between 18 and 24 months to complete a full cycle.[6]
The glycogen generation capacity is entirely dependent on, and determined by, the operating levels of the mitochondria in all of the cells of the human body;[8] however, the relation between the energy generated by the mitochondria and the glycogen capacity is very loose and is mediated by many biochemical pathways.[5] The energy output of full healthy mitochondrial function can be predicted exactly by a complicated theoretical argument, but this argument is not straightforward, as most energy is consumed by the brain and is not easily measurable.
Treatments
Although research is ongoing, treatment options are currently limited; vitamins are frequently prescribed, though the evidence for their effectiveness is limited.[9] Membrane penetrating antioxidants, such as the mitochondria-targeted antioxidant MitoQ (mitoquinol mesylate) have the most important role in improving mitochondrial dysfunction. Pyruvate has been proposed recently as a treatment option.[10] N acetylcysteine reverses many models of mitochondrial dysfunction.[11]
Spindle transfer, where the nuclear DNA is transferred to another healthy egg cell leaving the defective mitochondrial DNA behind, is a potential treatment procedure that has been successfully carried out on monkeys.[12] [13] Using a similar pronuclear transfer technique, researchers at Newcastle University led by Douglass Turnbull successfully transplanted healthy DNA in human eggs from women with mitochondrial disease into the eggs of women donors who were unaffected.[14][15] In such cases, ethical questions have been raised regarding biological motherhood, since the child receives genes and gene regulatory molecules from two different women. Using genetic engineering in attempts to produce babies free of mitochondrial disease is controversial in some circles and raises important ethical issues.[16][17] A male baby was born in Mexico in 2016 from a mother with Leigh syndrome using spindle transfer.[18]
In September 2012 a public consultation was launched in the UK to explore the ethical issues involved.[19] Human genetic engineering was used on a small scale to allow infertile women with genetic defects in their mitochondria to have children.[20] In June 2013, the United Kingdom government agreed to develop legislation that would legalize the 'three-person IVF' procedure as a treatment to fix or eliminate mitochondrial diseases that are passed on from mother to child. The procedure could be offered from 29 October 2015 once regulations had been established.[21][22][23] Embryonic mitochondrial transplant and protofection have been proposed as a possible treatment for inherited mitochondrial disease, and allotopic expression of mitochondrial proteins as a radical treatment for mtDNA mutation load.
Currently, human clinical trials are underway at GenSight Biologics (ClinicalTrials.gov # NCT02064569) and the University of Miami (ClinicalTrials.gov # NCT02161380) to examine the safety and efficacy of mitochondrial gene therapy in Leber's hereditary optic neuropathy.
Epidemiology
About 1 in 4,000 children in the United States will develop mitochondrial disease by the age of 10 years. Up to 4,000 children per year in the US are born with a type of mitochondrial disease.[24] Because mitochondrial disorders contain many variations and subsets, some particular mitochondrial disorders are very rare.
The average number of births per year among women at risk for transmitting mtDNA disease is estimated to approximately 150 in the United Kingdom and 800 in the United States.[25]
Notable cases
Notable people who suffered from mitochondrial disease include:
- Mattie Stepanek suffered from Dysautonomic Mitochondrial Myopathy. He was a poet, peace advocate, and motivational speaker who died at age 13.
- Rocco Baldelli is a coach and former center fielder in Major League Baseball who had to retire from active play at age 29 due to Mitochondrial Channelopathy.
References
- ↑ Salvatore DiMauro; Guido Davidzon (2005). "Mitochondrial DNA and disease" (PDF). Department of Neurology, Columbia University Medical Center. Retrieved March 20, 2013.
- ↑ Finsterer J (2007). "Hematological manifestations of primary mitochondrial disorders". Acta Haematol. 118 (2): 88–98. doi:10.1159/000105676. PMID 17637511.
- ↑ Scharfe C, Lu HH, Neuenburg JK, Allen EA, Li GC, Klopstock T, Cowan TM, Enns GM, Davis RW (2009). Rzhetsky, Andrey, ed. "Mapping gene associations in human mitochondria using clinical disease phenotypes". PLoS Comput Biol. 5 (4): e1000374. doi:10.1371/journal.pcbi.1000374. PMC 2668170
 . PMID 19390613.
. PMID 19390613. - ↑ Petersen; et al. (2004). "Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes". New England Journal of Medicine. Retrieved March 7, 2014.
- 1 2 Mitchell, Peter. "David Keilin`s respiratory chain concept and its chemiosmotic consequences" (PDF). Nobel institute.
- 1 2 Michelakis, Evangelos. "A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth". University of Alberta. University of Alberta, 2007.
- ↑ Lorini & Ciman, M, & M. "Hypoglycaemic action of Diisopropylammonium salts in experimental diabetes". Institute of Biochemistry, University of Padua, September 1962. Biochemical Pharmacology. 11: 823–827. doi:10.1016/0006-2952(62)90177-6.
- ↑ Stacpoole, Peter W (1998). "Clinical pharmacology and toxicology of dichloroacetate". University of Florida. Environmental health perspectives. pp. 989–994. PMC 1533324.
- ↑ Marriage B, Clandinin MT, Glerum DM (2003). "Nutritional cofactor treatment in mitochondrial disorders". J Am Diet Assoc. 103 (8): 1029–38. doi:10.1016/S0002-8223(03)00476-0. PMID 12891154.
- ↑ Tanaka M, Nishigaki Y, Fuku N, Ibi T, Sahashi K, Koga Y (2007). "Therapeutic potential of pyruvate therapy for mitochondrial diseases". Mitochondrion. 7 (6): 399–401. doi:10.1016/j.mito.2007.07.002. PMID 17881297.
- ↑ Frantz MC, Wipf P. Mitochondria as a target in treatment. Environ Mol Mutagen. 2010 Jun;51(5):462-75. doi:10.1002/em.20554
- ↑ Genetic advance raises IVF hopes By Pallab Ghosh. BBC News, science correspondent. Page last updated at 17:04 GMT, Wednesday, 26 August 2009 18:04 UK
- ↑ Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, Li Y, Ramsey C, Kolotushkina O, Mitalipov S (September 2009). "Mitochondrial gene replacement in primate offspring and embryonic stem cells". Nature. 461 (7262): 367–372. doi:10.1038/nature08368. PMC 2774772. PMID 19710649.
- ↑ Boseley, Sarah (2010-04-14). "Scientists reveal gene-swapping technique to thwart inherited diseases". London: Guardian.
- ↑ Craven, Lyndsey; Tuppen, Helen A.; Greggains, Gareth D.; Harbottle, Stephen J.; Murphy, Julie L.; Cree, Lynsey M.; Murdoch, Alison P.; Chinnery, Patrick F.; Taylor, Robert W.; Lightowlers, Robert N.; Herbert, Mary; Turnbull, Douglass M. (2010). "Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease". Nature. 465 (7294): 82–85. doi:10.1038/nature08958. PMC 2875160. PMID 20393463.

- ↑ "UK urged to permit IVF procedure to prevent fatal genetic diseases". London: The Guardian. 2015-04-30.
- ↑ "Three parent baby law is 'irresponsible' says Church of England ahead of vote". London: The Telegraph. 2015-04-30.
- ↑ Hamzelou, Jessica (2016-09-27). "Exclusive: World's first baby born with new "3 parent" technique". New Scientist. Retrieved 2016-11-26.
- ↑ Sample, Ian (2012-09-17). "Regulator to consult public over plans for new fertility treatments". The Guardian. London. Retrieved 8 October 2012.
- ↑ "Genetically altered babies born". BBC News. 2001-05-04. Retrieved 2008-04-26.
- ↑ The Human Fertilisation and Embryology (Mitochondrial Donation) Regulations 2015 No. 572
- ↑ "UK government backs three-person IVF". BBC News. 27 June 2013.
- ↑ Knapton, Sarah (1 March 2014) 'Three-parent babies' could be born in Britain next year The Daily Telegraph Science News, Retrieved 1 March 2014
- ↑ The Mitochondrial and Metabolic Disease Center
- ↑ Gorman, Gráinne S.; Grady, John P.; Ng, Yi; Schaefer, Andrew M.; McNally, Richard J.; Chinnery, Patrick F.; Yu-Wai-Man, Patrick; Herbert, Mary; Taylor, Robert W.; McFarland, Robert; Turnbull, Doug M. (2015). "Mitochondrial Donation — How Many Women Could Benefit?". New England Journal of Medicine. 372: 150130091413004. doi:10.1056/NEJMc1500960. ISSN 0028-4793.
External links
| Wikimedia Commons has media related to Mitochondrial diseases. |
- Mitochondrial disease at DMOZ
- North American Mitochondrial Disease Consortium
- Mitochondrial Disease Action Committee