Genome engineering
Genome engineering refers to the strategies and techniques developed for the targeted, specific modification of the genetic information – or genome – of living organisms.
It represents a very active field of research because of the wide range of possible applications, particularly in the areas of human health - the correction of a gene carrying a harmful mutation, the production of therapeutic proteins, the elimination of persistent viral sequences - agricultural biotechnology - the development of new generations of genetically modified plants - and for the development of research tools - for example, to explore the function of a gene.
Early technologies developed to insert a gene into a living cell, such as transgenesis, are limited by the random nature of the insertion of the new sequence into the genome. The new gene is positioned blindly, and may inactivate or disturb the functioning of other genes or even cause severe unwanted effects; it may trigger a process of cancerization, for example. Furthermore, these technologies offer no degree of reproducibility, as there is no guarantee that the new sequence will be inserted at the same place in two different cells.
The major advantage of genome engineering, which uses more recent knowledge and technology, is that it enables a specific area of the DNA to be modified, thereby increasing the precision of the correction or insertion, preventing any cell toxicity and offering perfect reproducibility.
Genome engineering and synthetic genomics (designing artificial genomes) are currently among the most promising technologies in terms of applied biological research and industrial innovation.
General principles
Early approaches to genome engineering involved modifying genetic sequences using only homologous recombination. Using a homologous sequence located on another strand as a model can lead this natural DNA maintenance mechanism to repair a DNA strand. It is possible to induce homologous recombinations between a cellular DNA strand and an exogenous DNA strand inserted in the cell by researchers, using a vector such as the modified genome of a retrovirus. The recombination phenomenon is flexible enough for a certain level of change (addition, suppression or modification of a DNA portion) to be introduced to the targeted homologous area.
In the 1980s, Mario R. Capecchi and Oliver Smithies worked on the homologous recombination of DNA as a “gene targeting” tool; in other words, as an instrument for the inactivation or modification of specific genes. Working with Martin J. Evans, they developed a process for the modification of the mouse genome by modifying the DNA of mouse embryonic stem cells in culture and injecting these modified stem cells into mouse embryos. Genetically modified mice generated using this method make useful laboratory models to study human diseases. This tool is now commonly used in medical research. The three researchers were awarded the 2007 Nobel Prize in Medicine for their work.[1]
Modifying genomes using only homologous recombination remained a long and random process until additional developments were made that could increase the rate of homologous recombination in somatic cell types. These developments include two mechanistically distinct methods of triggering the cells inherent DNA repair mechanisms which are required to insert a foreign gene sequence into a live cell. The first such methods use site-directed endonucleases (restriction enzymes), such as zinc finger nucleases (ZFNs), meganucleases and transcription activator like effector nucleases (TALENs). Site directed endonucleases achieve gene modification through causing double stranded DNA (dsDNA) breaks which triggers the cells natural DNA repair mechanism, predominantly non homologous end joining (NHEJ) as well as a low frequency of homologous recombination (HR). The second method is recombinant adeno-associated virus (rAAV) mediated genome engineering which induces high frequencies of homologous recombination alone, thus forgoing the need to perform dsDNA breaks.
Methods in genome engineering:
- Insertion involves introducing a gene into a chromosome to obtain a new function (for example to obtain a better drought-resistant plant) or to compensate for a defective gene, particularly by making it possible to manufacture a functional protein if the protein produced by the patient is defective (such as factor VIII in hemophilia A).
- Inactivation, or “knock-out”, is today mainly used in fundamental research to shed light on the function of a gene by observing the anomalies that occur as a result of its inactivation. It can also have other applications, for example to remove a persistent viral sequence from infected cells, or in agriculture to eliminate the irritant or allergenic properties of a plant.
- Correction aims to remove and replace a defective gene sequence with a functional sequence. This correction can be performed on a very short sequence, sometimes just a few nucleotides, such as in the case of drepanocytosis (sickle cell anemia). In plants, this manipulation can also help improve the properties of a species without the addition of foreign DNA.
Multiplex Automated Genomic Engineering (MAGE)
The methods for scientists and researchers wanting to study genomic diversity and all possible associated phenotypes were very slow, expensive, and inefficient. Prior to this new revolution, researchers would have to do single-gene manipulations and tweak the genome one little section at a time, observe the phenotype, and start the process over with a different single-gene manipulation.[2] Therefore, researchers at the Wyss Institute at Harvard University designed the MAGE, a powerful technology that improves the process of in vivo genome editing. It allows for quick and efficient manipulations of a genome, all happening in a machine small enough to put on top of a small kitchen table. Those mutations combine with the variation that naturally occurs during cell mitosis creating billions of cellular mutations.
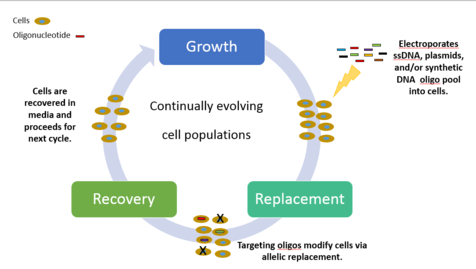
Chemically combined, synthetic single-stranded DNA (ssDNA) and a pool of oligionucleotides are introduced at targeted areas of the cell thereby creating genetic modifications. The cyclical process involves transformation of ssDNA (by electroporation) followed by outgrowth, during which bacteriophage homologous recombination proteins mediate annealing of ssDNAs to their genomic targets. Experiments targeting selective phenotypic markers are screened and identified by plating the cells on differential medias. Each cycle ultimately takes 2.5 hours to process, with additional time required to grow isogenic cultures and characterize mutations. By iteratively introducing libraries of mutagenic ssDNAs targeting multiple sites, MAGE can generate combinatorial genetic diversity in a cell population. There can be up to 50 genome edits, from single nucleotide base pairs to whole genome or gene networks simultaneously with results in a matter of days.[2]
MAGE experiments can be divided into three classes, characterized by varying degrees of scale and complexity: (i) many target sites, single genetic mutations; (ii) single target site, many genetic mutations; and (iii) many target sites, many genetic mutations.[2] An example of class three was reflected in 2009, where Church and colleagues were able to program Escherichia coli to produce five times the normal amount of lycopene, an antioxidant normally found in tomato seeds and linked to anti-cancer properties. They applied MAGE to optimize the 1-deoxy-d-xylulose-5-phosphate (DXP) metabolic pathway in Escherichia coli to overproduce isoprenoid lycopene. It took them about 3 days and just over $1,000 in materials. The ease, speed, and cost efficiency in which MAGE can alter genomes can transform how industries approach the manufacturing and production of important compounds in the bioengineering, bioenergy, biomedical engineering, synthetic biology, pharmaceutical, agricultural, and chemical industries.
Transfection by causing dsDNA breaks
Researchers wishing to efficiently eliminate a gene to study the resulting loss of its function are increasingly opting for a “molecular scissors” approach. These are enzymes with specific properties which enable them to cut the long double DNA strand along the sequence to be modified, thereby triggering the NHEJ and HR process at the required location.
Restriction enzymes commonly used in molecular biology to cut DNA interact with sequences of 1 to 10 nucleotides. These sequences, which are very short and generally palindromic, often occur at several sites in the genome (the human genome comprises 6.4 billion bases). Restriction enzymes are therefore likely to cut the DNA molecule several times. In their efforts to find a genome surgery approach offering a higher degree of accuracy and security, scientists therefore turned to more precise tools.
More targeted genome engineering can be performed by using enzymes which are able to recognize and interact with DNA sequences that are sufficiently long so as to occur only once, with high probability, in any given genome. The DNA modification therefore takes place precisely at the site of the target sequence. Because their recognition sites are larger than 12 base pairs, meganucleases, zinc finger nucleases, and TALEN fusions offer this degree of precision.
Once the DNA has been cut, natural DNA repair mechanisms and homologous recombination enable the incorporation of a modified sequence or a new gene.
The success of these different stages (recognition, cleavage and recombination) depends on various factors, including the efficacy of the vector that introduces the enzyme into the cell, the enzyme cleavage activity, the cell’s capacity for homologous recombination and probably the state of the chromatin at the given locus.
Meganuclease-based Engineering
Meganucleases, discovered in the late 1980s, are enzymes in the endonuclease family which are characterized by their capacity to recognize and cut large DNA sequences (from 12 to 40 base pairs).[3] The most widespread and best known meganucleases are the proteins in the LAGLIDADG family, which owe their name to a conserved amino acid sequence.
These enzymes were identified in the 1990s as promising tools for genome engineering. However, even though they occur in nature and each one exhibits slight variations in its DNA recognition site, there is virtually no chance of finding the exact meganuclease required to act on a specific DNA sequence. Each new genome engineering target therefore requires an initial protein engineering stage to produce a custom meganuclease.
Two methods for creating custom meganucleases:
- Mutagenesis involves generating collections of variants using a meganuclease with properties similar to the desired enzyme, then selecting these variants using high-throughput screening. This procedure can be optimized by adopting what are known as "semi-rational" methods, in which the structural data is electronically processed in order to focus the mutagenesis to the part of the enzyme that interacts with DNA and triggers the cleavage.[4]
- Combinatorial assembly is a method whereby protein subunits from different enzymes can be associated or fused.[5]
These two approaches can be combined.
A large bank containing several tens of thousands of protein units has been created. These units can be combined to obtain chimeric meganucleases that recognize the target site, thereby providing research and development tools that meet a wide range of needs (fundamental research, health, agriculture, industry, energy, etc.).
This technique has enabled the development of several meganucleases specific for sequences in the genomes of viruses, plants, etc., and the industrial-scale production of two meganucleases able to cleave the human XPC gene; mutations in this gene result in Xeroderma pigmentosum, a severe monogenic disorder that predisposes the patients to skin cancer and burns whenever their skin is exposed to UV rays.[6]
Another approach involves using computer models to try to predict as accurately as possible the activity of the modified meganucleases and the specificity of the recognized nucleic sequence.[7] The Northwest Genome Engineering Consortium, a US consortium funded by the National Institutes of Health, has adopted this approach with the aim of treating leukemia by modifying hematopoietic stem cells. The model’s prediction has been verified and guided by means of directed mutagenesis and in vitro biochemical analysis.
A third approach has been taken by the American biotechnology company Precision Biosciences, Inc. The company, funded by the National Institutes of Health and the National Institute of Standards and Technology, has developed a fully rational design process called the Directed Nuclease Editor (DNE) which is capable of creating highly specific engineered meganucleases that successfully target and modify a user-defined location in a genome.[8]
Zinc finger nuclease-based Engineering
Zinc finger motifs occur in several transcription factors. The zinc ion, found in 8% of all human proteins, plays an important role in the organization of their three-dimensional structure. In transcription factors, it is most often located at the protein-DNA interaction sites, where it stabilizes the motif. The C-terminal part of each finger is responsible for the specific recognition of the DNA sequence.
The recognized sequences are short, made up of around 3 base pairs, but by combining 6 to 8 zinc fingers whose recognition sites have been characterized, it is possible to obtain specific proteins for sequences of around 20 base pairs. It is therefore possible to control the expression of a specific gene. It has been demonstrated that this strategy can be used to promote a process of angiogenesis in animals.[9] It is also possible to fuse a protein constructed in this way with the catalytic domain of an endonuclease in order to induce a targeted DNA break, and therefore to use these proteins as genome engineering tools.[10]
The method generally adopted for this involves associating two proteins – each containing 3 to 6 specifically chosen zinc fingers – with the catalytic domain of the FokI endonuclease. The two proteins recognize two DNA sequences that are a few nucleotides apart. Linking the two zinc finger proteins to their respective sequences brings the two endonucleases associated with them closer together. This means that they can be dimerized and then cut the DNA molecule.
Several approaches are used to design specific zinc finger nucleases for the chosen sequences. The most widespread involves combining zinc-finger units with known specificities (modular assembly). Various selection techniques, using bacteria, yeast or mammal cells have been developed to identify the combinations that offer the best specificity and the best cell tolerance. Although the direct genome-wide characterization of zinc finger nuclease activity has not been reported, an assay that measures the total number of double-strand DNA breaks in cells found that only one to two such breaks occur above background in cells treated with zinc finger nucleases with a 24 bp composite recognition site and obligate heterodimer FokI nuclease domains.[11]
Zinc finger nucleases are research and development tools that have already been used to modify a range of genomes, in particular by the laboratories in the Zinc Finger Consortium. The US company Sangamo BioSciences uses zinc finger nucleases to carry out research into the genetic engineering of stem cells and the modification of immune cells for therapeutic purposes.[12][13] Modified T lymphocytes are currently undergoing phase I clinical trials to treat a type of brain tumor (glioblastoma) and in the fight against AIDS.[11]
TALEN

Transcription activator-like effector nucleases (TALENs) are artificial restriction enzymes generated by fusing a specific DNA-binding domain to a non-specific DNA cleaving domain. The DNA binding domains, which can be designed to bind any desired DNA sequence, comes from TAL effectors, DNA-binding proteins excreted by plant pathogenic Xanthomanos app. Tal effectors consists of repeated domains, each which contains a highly considered sequence of 34 amino acids, and recognize a single DNA nucleotide. The nuclease can create double strand breaks at the target site that can be repaired by error-prone non-homologous end-joining (NHEJ), resulting in gene disruptions through the introduction of small insertions or deletions. TALEN constructs are used in a similar way to designed zinc finger nucleases, and have three advantages in targeted mutagenesis: (1) DNA binding specificity is higher, (2) off-target effects are lower, and (3) construction of DNA-binding domains is easier.
CRISPRs
CRISPRs (Clustered Regularly Interspaced Short Palindromic Repeats) are genetic elements that bacteria use as a kind of acquired immunity to protect against viruses. They consist of short sequences that originate from viral genomes and have been incorporated into the bacterial genome. Cas (CRISPR associated proteins) process these sequences and cut matching viral DNA sequences. By introducing plasmids containing Cas genes and specifically constructed CRISPRs into eukaryotic cells, the eukaryotic genome can be cut at any desired position.[14] Several companies, including Editas, have been working to monetize the CRISPR method while developing gene-specific therapies.[15][16]
Homologous recombination
rAAV-stimulated homologous recombination
Recombinant adeno-associated virus (rAAV) mediated genome engineering alters genomic DNA by stimulating pre-established and differentiated human cell lines by homologous recombination, which is otherwise low in these cells.[17][18] Adeno-associated viruses have high transduction rates and have a unique property of stimulating endogenous homologous recombination. The rAAV vector alters a specific genomic locus with a supplied replacement sequence by recombination without causing double strand DNA breaks.[17]
See also
- Gene drive
- Synthetic biology
- Recombinant AAV mediated genome engineering
- Isogenic human disease models
- homologous recombination
- Genome editing with engineered nucleases
- Biological engineering
- Homing endonuclease
- Protein engineering
- Protein design
References
- ↑ "The Nobel Prize in Physiology or Medicine 2007". www.nobelprize.org. Retrieved 2016-05-15.
- 1 2 3 Gallagher, Ryan R; Li, Zhe; Lewis, Aaron O; Isaacs, Farren J (2014-01-01). "Rapid editing and evolution of bacterial genomes using libraries of synthetic DNA". Nature Protocols. 9 (10): 2301–2316. doi:10.1038/nprot.2014.082.
- ↑ Stoddard, BL (2006). "Homing endonuclease structure and function". Quarterly Reviews in Biophysics. 38 (1): 49–95. doi:10.1017/s0033583505004063.
- ↑ Seligman, LM; Chisholm, KM; Chevlier, BS; Chadsey, MS; Edward, ST; Savage, JH; Veillet, AL (2002). "Mutations altering the cleavage specificity of a homing endonuclease". Nucleic Acids Research. 30: 3870–3879. doi:10.1093/nar/gkf495. PMC 137417
 . PMID 12202772.
. PMID 12202772. - ↑ Arnould, S; Chams, P; Perez, C; Lacroix, E; Duclert, A; Epinat, JC; Stricher, F; Petit, AS; Patin, A; Guillier, S; Rolland, S; Prieto, J; Blanco, FJ; Bravo, J; Montaya, G; Serrano, L; Duchateau, P; Pâques, F (2006). "Engineering of large numbers of highly specific homing endonucleases that induce recombination to novel DNA targets". Journal of Molecular Biology. 355: 443–458. doi:10.1016/j.jmb.2005.10.065. PMID 16310802.
- ↑ Redondo P, Prieto J, Munoz IG, Alibés A, Stricher F, Serrano L, Cabaniols J-P, Daboussi F, Arnould S, Perez C, Duchateau P, Pâques F, Blanco FJ, Montoya G (2008). Molecular basis of xeroderma pigmentosum group C DNA recognition by engineered meganucleases" Nature 456(7218): 107-111.
- ↑ Ashworth, J; Taylor, GK; Havranek, JJ; Quadri, SA; Stoddard, BL; Baker, D (2010). "Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs". Nucleic Acids Research. 38 (16): 5601–5608. doi:10.1093/nar/gkq283.
- ↑ Gao, Huirong; Smith, James; Yang, Maizhu; Jones, Spencer; Stagg, Jessice; Djukanvic, Vesna; Nicholson, Mike; West, Ande; Bidney, Dennis; Falco, Carl; Jantz, Derek; Lyznik, L. Alexander (January 2010). "Heritable Targeted Mutagenesis in Maize Using a Dedicated Meganuclease". The Plant Journal. 61 (1): 176–87. doi:10.1111/j.1365-313X.2009.04041.x. PMID 19811621.
- ↑ Rebar, EJ; Huang, Y; Hickey, R; Nath, AK; Meoli, D; Nath, S; Chen, B; Xu, L; Liang, Y; Jamieson, AC; Zhang, L; Spratt, SK; Case, CC; Wolfe, A; Giordano, FJ (2002). "Induction of angiogenesis in a mouse model using engineering transcription factors". Nature Medicine. 8: 1427–1432. doi:10.1038/nm1202-795.
- ↑ Kim Kim, H-G; Cha, J; Chandrasegaran (2007). "Hybrid restriction enzymes : Zinc finger fusions to Fok I cleavage domain". Proceedings of the National Academy of Sciences of the United States of America. 93: 1156–1160.
- 1 2 Urnov, FD; Rebar, EJ; Holmes, MC; Zang, HS; Grogory, PD (2010). "Genome editing with engineered zinc finger nucleases". Nature Reviews. 11: 636–646. doi:10.1038/nrg2842. PMID 20717154.
- ↑ Reik, A; et al. (2008). "Zinc finger nucleases targeting the glucocorticoid receptor allow IL-13 zetakine transgenic CTLs to kill glioblastoma cells in vivo in the presence of immunosuppressing glucocorticoids". Mol. Ther. 16: S13–S14.
- ↑ Holt, N; et al. (2010). "Human hematopoitic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo". Nature Biotechnology. 28: 839–847. doi:10.1038/nbt.1663.
- ↑ Young, Susan (11 February 2014) Genome Surgery MIT Technology Review, Retrieved 17 February 2014
- ↑ Fye, Shaan. "Genetic Rough Draft: Editas and CRISPR". The Atlas Business Journal. Retrieved 19 January 2016.
- ↑ Regalado, Antonio (2015-11-05). "CRISPR Gene Editing to Be Tested on People by 2017, Says Editas". MIT Technology Review. Retrieved 2016-06-21.
- 1 2 Facile methods for generating human somatic cell gene knockouts using AAV Nucleic Acids Res. 2 January 2004; 32(1): e3
- ↑ Improved methods for the generation of human gene knockout and knock-in cell lines Nucleic Acids Res. 7 October 2005; 33(18): e158