Fluoxymesterone
 | |
| Clinical data | |
|---|---|
| Trade names | Halotestin, Ultandren |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682690 |
| Pregnancy category |
|
| Routes of administration | Oral |
| ATC code | G03BA01 (WHO) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% Oral |
| Metabolism | Hepatic |
| Biological half-life | 9.5 hours |
| Excretion | urine |
| Identifiers | |
| |
| CAS Number |
76-43-7 |
| PubChem (CID) | 6446 |
| IUPHAR/BPS | 2861 |
| DrugBank |
DB01185 |
| ChemSpider |
6205 |
| UNII |
9JU12S4YFY |
| KEGG |
D00327 |
| ChEBI |
CHEBI:5120 |
| ChEMBL |
CHEMBL1445 |
| Chemical and physical data | |
| Formula | C20H29FO3 |
| Molar mass | 336.441 g/mol |
| 3D model (Jmol) | Interactive image |
| |
| |
| (verify) | |
Fluoxymesterone (brand names Halotestin, Ultandren), also known as 9α-fluoro-11β-hydroxy-17α-methyltestosterone, is a synthetic, orally active androgenic-anabolic steroid (AAS) and 17α-methylated derivative of testosterone with strong androgenic activity that has been used in the treatment of hypogonadism and delayed puberty in males and breast cancer in women. It is approximately five times as potent as an AAS as testosterone.[1] The antitumor activity of fluoxymesterone appears related to reduction or competitive inhibition of prolactin receptors or estrogen receptors or production. Dissimilar to testosterone, fluoxymesterone has a 100% bioavailability, as the methylation of the C17α-position in fluoxymesterone inhibits hepatic metabolism by enzymatic oxidation of 17β-hydroxyl, allowing its absorption into the bloodstream for transport around the body. While there are many legitimate pharmaceutical applications of fluoxymesterone, it is also abused, leading to the development of analytical techniques by which fluoxymesterone doping can be identified.
Pharmacology
Like many C17α-alkylated steroids, fluoxymesterone has relatively low affinity for the androgen receptor (AR). Even so, its actions are mediated by the AR, most likely due to its prolonged elimination half-life of approximately 9.2 hours.[2]
Fluoxymesterone has been found to act as a potent inhibitor of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) (IC50 = 60–630 nM), with a potency comparable to that of the 11β-HSD2 inhibitor glycyrrhetinic acid.[3][4] This unique action of fluoxymesterone is likely related to its 11β-hydroxyl group.[3] 11β-HSD2 is responsible for the inactivation of the glucocorticoids cortisol and corticosterone (into cortisone and 11-dehydrocorticosterone, respectively).[3][4] Inhibition of 11β-HSD2 by fluoxymesterone may result in mineralocorticoid receptor overactivation and associated side effects such as hypertension and fluid retention, and has been hypothesized to be involved in the cardiovascular and other adverse effects of fluoxymesterone.[3][4]
Chemistry
Synthesis
Step One: The first step in the synthesis of fluoxymesterone is the microbiological oxidation of commercially available androstenedione (1.11) by Actinomyces; this introduces a hydroxyl group to the 11α-position (1.12), which is then oxidised to a ketone using Jones’ reagent, yielding the 3,11,17-triketone, adrenosterone (1.13). Pyrrolidine then reacts to form an enamine (1.14) by reaction with the 3α-keto group, protecting it from alkylation in a subsequent step. The regioselectivity of pyrrolidine for reaction at the 3α-position occurs inherently in the structure of adrenosterone, due to the position of the sterically bulky methyl groups. In subsequent steps, alkylation of the 17-keto group (1.14) using Grignard reagent, addition of hydride at the 11-position (1.15) and regeneration of the protected 3-keto group yields the starting material (1.16) for the final steps of the fluoxymesterone synthesis. This involves more standard synthetic transformations.
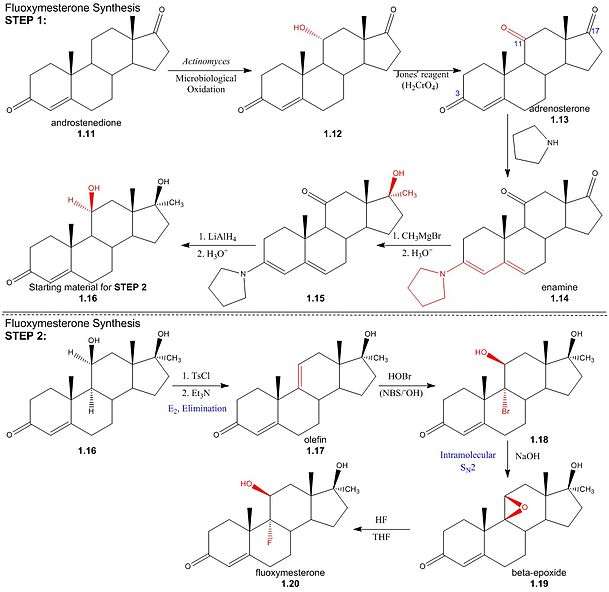
StepTwo: The 11α-hydroxyl of the starting material (1.16) is sulfonylated by p-toluenesulfonyl chloride; addition of trimethylamine (base) deprotonates the 11α-carbon, yielding an (E2) elimination of tosylate (pka - 5) to give olefin (1.17). Stereospecificity of reaction between olefin and hypobromous acid (HOBr) in base, N-bromosuccinimide (NBS), is determined by the formation of a bromonium intermediate; the electrophilic bromonium cation approaches the ring’s less sterically hindered α-face and is attacked by the π-electron density of the alkene. The hydroxide ion then attacks from above the ring (β-face) at the 11-carbon, resulting in a structure (1.18) by the stereospecific addition of hydroxyl and bromine across the double bond. Addition of sodium hydroxide results in deprotonation of the 11α-hydroxyl, and the subsequent structure undergoes an intramolecular SN2 epoxy ring formation. The epoxy ring of the β-epoxide (1.19) is protonated to give an oxironium ion intermediate. In a concerted process, fluoride attacks the ring’s α-face from below, as one of the two oxygen-carbon bonds is broken on the opposite face; hence regenerating the 11α-hydroxyl trans to the fluorine substituent. The resulting structure (1.20) is the androgenic steroid, fluoxymesterone.
Detection in body fluids
Detection of halotestin and other such illegal anabolic steroids in sports is achieved by GS-MS identification of urinary excreted anabolic steroids and their metabolites. In a test for halotestin, a dry residue obtained from a urine sample is dissolved in dimethylformamide and a sulfur trioxide-pyridine complex and is heated with 1% potassium carbonate solution. Halotestin and many of its metabolites contain two polar hydroxyl groups, leading to intermolecular hydrogen bonding that increases their boiling point and reduces volatility. In order to attain a gaseous sample for GC-MS, the products of hydrolysis are extracted, dissolved in methanol and derivatised to form volatile trimethylsilyl (TMS) esters by adding N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA) and trimethylsilylimidazole (TMSImi).[5]
References
- ↑ Dr. K.V. Sastry (2008). Endocrinology and Reproductive Biology. Page 150. ISBN 81-7133-777-5.
- ↑ Seth Roberts (2009). Anabolic Pharmacology.
- 1 2 3 4 Fürstenberger C, Vuorinen A, Da Cunha T, Kratschmar DV, Saugy M, Schuster D, Odermatt A (2012). "The anabolic androgenic steroid fluoxymesterone inhibits 11β-hydroxysteroid dehydrogenase 2-dependent glucocorticoid inactivation". Toxicol. Sci. 126 (2): 353–61. doi:10.1093/toxsci/kfs022. PMID 22273746.
- 1 2 3 Joseph JF, Parr MK (2015). "Synthetic androgens as designer supplements". Curr Neuropharmacol. 13 (1): 89–100. doi:10.2174/1570159X13666141210224756. PMC 4462045
 . PMID 26074745.
. PMID 26074745. - ↑ Schänzer, Willi; Opfermann, Georg; Donike, Manfred (1992-11-01). "17-Epimerization of 17α-methyl anabolic steroids in humans: metabolism and synthesis of 17α-hydroxy-17β-methyl steroids". Steroids. 57 (11): 537–550. doi:10.1016/0039-128X(92)90023-3.
Further reading
- Daniels, R. C. (February 1, 2003). The Anabolic Steroid Handbook. Richard C Daniels. p. 80. ISBN 0-9548227-0-6.