Fleming–Tamao oxidation
| Fleming-Tamao oxidation | |
|---|---|
| Named after | Ian Fleming Kohei Tamao |
| Reaction type | Organic redox reaction |
| Identifiers | |
| Organic Chemistry Portal | fleming-tamao-oxidation |
| RSC ontology ID | RXNO:0000210 |
The Fleming–Tamao oxidation, or Tamao–Kumada–Fleming oxidation, converts a carbon–silicon bond to a carbon–oxygen bond with a peroxy acid or hydrogen peroxide. Fleming–Tamao oxidation refers to two slightly different conditions developed concurrently in the early 1980s by the Kohei Tamao and Ian Fleming research groups.[1][2][3]

The reaction is stereospecific with retention of configuration at the carbon–silicon bond.[2][3] This allows the silicon group to be used as a functional equivalent of the hydroxyl group. Another key feature of the silicon group is that it is relatively stable due to the presence of the silicon atom, and therefore can tolerate various reaction conditions that the hydroxyl group can not tolerate. Due to the stability of the silicon group, organosilicon compounds are useful in the total synthesis of complex natural products and pharmaceutical drugs. For instance, the Fleming–Tamao oxidation has been used to accomplish the synthesis of subunits of tautomycin,[4] an inhibitor that is used as a lead cancer compound and as an immunosuppressant.
History
In 1983, Tamao and co-workers were the first to report the successful transformation of an allyl alkoxy silyl to an allyl alcohol without an allylic shift.[5] In their report, the chemists observed that the hydroxyl group was introduced exclusively onto the carbon atom to which the silicon atom was attached. In the same year, Tamao and group published another paper that showed that the carbon–silicon bond in alkoxy organosilicon compounds can be cleaved using H2O2 or m-CPBA under acidic, basic (chemistry), or neutral conditions, to afford the corresponding alcohols.[6] A year later, Ian Fleming and group reported that the dimethylphenylsilyl (Me2PhSi) group can be converted to an hydroxyl group in a two-pot sequence.[2] Later, in 1987, Fleming reported a one-pot variant to the two-pot sequence in which either bromine or mercuric ion acts as the electrophile. These early findings paved the way for the development of a large number of silicon-based reagents and the use of various silyl groups as functional equivalents of the hydroxyl group.
Mechanisms
Tamao–Kumada oxidation
Although the mechanism below is for the basic condition, the proposed mechanism[1][7] for the Tamao oxidation is similar under each condition. The mechanism below contains at least one fluorine atom as the substituent, which is the prototype structure that Tamao studied. Fluoride, provided by a fluoride source or a donor solvent, attacks the fluorosilane in a fast and reversible step to give a pentaco-ordinated species. This species is more electrophilic than the fluorosilane, thereby promoting attack by the nucleophilic oxidant to yield the negatively charged hexaco-ordinated transition state. This step was determined to be the rate determining step based on kinetic studies done by Tamao.[8] Further studies by Tamao on the steric and electronic effects of different groups attached to the silicon led him to suggest that attack by the oxidant trans to the electronegative fluoride group is energetically favored. The group cis to the peroxide oxygen in the transition state structure then migrates preferentially, thus explaining the retention of configuration at the carbon center. Finally, the new silicon–oxygen bond of the hexaco-ordinated species is hydrolyzed by water in the reaction medium. Subsequent workup produced the expected alcohol.

Fleming oxidation
Two-pot sequence
Unlike the Tamao oxidation whose starting material is an activated heteroatom-substituted silyl group, the Fleming oxidation utilizes a more robust silyl group which has only carbon atoms attached to the silicon atom. The prototype silyl structure that Fleming used was dimethylphenylsilyl. This aryl silane is then converted to the more reactive halo- or heterosilane to initiate the oxidation.[2][3] The mechanism of the two-pot sequence[1][7] differs from the Tamao oxidation since the reagents are different. First, an electrophile attacks the phenyl ring in the ipso position to give a beta-carbocation that is stabilized by the silicon group. A heteroatom then attacks the silicon group, which allows the phenyl ring to leave, in a key step referred to as protodesilylation of the arylsilane. The alkyl group undergoes 1,2 migration from the silicon to the oxygen atom. Aqueous acid mediated Hydrolysis and subsequent workup yield the desired alcohol. It is difficult to prevent small resulting silyl-alcohols from dehydrating to form siloxanes.

One-pot sequence
The main difference between the one-pot and two-pot sequences is that the former has bromine or mercuric ion as the electrophile that attacks the benzene ring. The bromine electrophile is generated by diatomic bromine or another source such as potassium bromide, which can be oxidized to generate bromine in situ by the peracetic acid. The source of the mercuric ion is mercuric acetate, and this reagent is mixed with peracetic acid in AcOH to provide the oxidizing conditions. The mechanism for the one-pot and two-pot sequences is the same since the bromine or mercuric ion attacks the phenyl ring instead of the hydrogen ion.[3][9]
Scope
The Tamao–Kumada oxidation, or the Tamao oxidation, uses a silyl group with a hydrogen atom, a heteroatom or an electron-donating group attached to the silicon atom to make it more reactive. Tamao used either fluorine or chlorine atom, or an alkoxy (OR) or amine group (NR2) as the substituent on the substrates.[5] In addition to varying the percent composition of oxidants and combining different solvents, Tamao also used additives such as acetic anhydride (Ac2O), potassium hydrogen fluoride (KHF2), and potassium hydrogen carbonate (KHCO3) or sodium hydrogen carbonate (NaHCO3) to make the reaction conditions slightly acidic, neutral, and alkaline, respectively. The different conditions were used to observe the effect that pH environment had on the oxidative cleavage of the various alkoxy groups. Below is an example of each reaction condition.
![{\displaystyle {\begin{matrix}{\text{Reaction conditions of Tamao oxidation}}\\{}\\{\ce {R}}{-}{\color {Blue}{\ce {Si}}}{\ce {R'_{2}X}}{\begin{cases}{\ce {->[30\%\ {\ce {H2O2,\ KHCO3\ or\ NaHCO3}}][{\ce {MeOH/THF,\ 60^{\circ }C}}]R}}{-}{\color {Blue}{\ce {O}}}{\ce {H}}&{\color {Blue}{\text{basic condtions}}}\\\\{\ce {->[30\%\ {\ce {H2O2,\ Ac2O,\ KHF2}}][{\text{DMF, r.t.}}]R}}{-}{\color {Blue}{\ce {O}}}{\ce {H}}&{\color {Blue}{\text{acidic conditions}}}\\\\{\ce {->[30\%\ {\ce {H2O2,\ KHF2}}][{\text{DMF, r.t. to }}60^{\circ }{\text{C}}]R}}{-}{\color {Blue}{\ce {O}}}{\ce {H}}&{\color {Blue}{\text{neutral conditions}}}\end{cases}}\\\\{\ce {X}}={\ce {H,F,Cl,OR,NR2}}\\{\ce {R'}}={\ce {X,Me,Et,iPr,Ph}}\\\end{matrix}}}](../I/m/1a97d70ed29cfa828e881bb136ee345c28c54a0e.svg)
Variations
Recently, the Fleming–Tamao oxidation has been used to generate phenol and substituted phenols in very good yield.[10]

The Tamao oxidation was used to synthesize acid, aldehyde, and ketone under varying reaction conditions.[11] Whereas the carbon-silicon bond of a substituted alkylsilyl is cleaved to a carbon-oxygen single bond, a substituted alkenylsilyl group is transformed to a carbonyl under the same Tamao oxidation conditions employed for alkylsilane.[11]

Advantages of a C–Si linkage
The silyl group is a non-polar and relatively unreactive species and is therefore tolerant of many reagents and reaction conditions that might be incompatible with free alcohols. Consequently, the silyl group also eliminates the need for introduction of hydroxyl protecting groups. In short, by deferring introduction of an alcohol to a late synthetic stage, opting instead to carry through a silane, a number of potential problems experienced in total syntheses can be mitigated or avoided entirely.[1]
Steric effects
One of the major pitfalls of either the Fleming or Tamao oxidations is steric hindrance.[2] Increasing the steric bulk at the silicon center generally slows down reaction, potentially even suppressing reaction entirely when certain substituents are employed. In general, less bulky groups such as methyl or ethyl favor oxidation, while bulkier groups such as tert-butyl slow down or stop oxidation. There are special cases in which this pattern in not followed. For example, alkoxy groups tend to enhance oxidation,[6] while oxidation does not proceed under normal conditions when three alkyl substituents are attached to the silicon atom. The trend below illustrates the order in which oxidation proceeds.
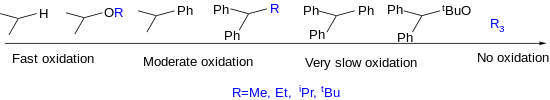
Applications
Natural product synthesis
The natural product, (+)− pramanicin, became an interesting target for synthesis because it was observed to be active against a fungal pathogen that resulted in meningitis in AIDS patients. Therefore, its synthesis[12] which utilized the Fleming–Tamao oxidation as a crucial step has been relevant to chemists as well as human beings who are inflicted by AIDS. The antifungal agent has also been shown previously to induce cell death and increase calcium levels in vascular endothelial cells. Furthermore, (+)– pramanicin has a wide range of potential applications against human diseases.

Polyol synthesis
Polyols and diols are especially useful to the food industry and polymer chemistry. Their importance is underscored by the fact that they can be used as sugar replacers for diabetics or those who choose to have sugar-free or low-calorie diets. The Fleming-Tamao has been applied in the synthesis of stereoselective diols. Woerpel[13] used the reaction to synthesize anti-1,3 diols from functionalized silyl anion.
Alternatively, Hara, K.; Moralee, and Ojima[14] achieved syn-1,3 diols using Tamao oxidation.
See also
External links
References
- 1 2 3 4 Jones, G.R.; Landais,Y. (1996), "The Oxidation of the Carbon-Silicon Bond", Tetrahedron, 52: 7599–7662, doi:10.1016/s0040-4020(96)00038-5
- 1 2 3 4 5 Fleming, I.; Henning, R.; Plaut, H.E. (1984), "The Phenyldimethylsilyl Group as a Masked Form of the Hydroxy Group", J. Chem. Soc., Chem. Commun.: 29–31, doi:10.1039/C39840000029
- 1 2 3 4 Fleming, I.; Henning, R.; Parker, D.C.; Plaut, H.E.; Sanderson, P.E.J. (1995), "The Phenyldimethylsilyl Group as a Masked Hydroxy Group", J. Chem. Soc., Perkin Trans. 1: 317–37, doi:10.1039/P19950000317
- ↑ James, M. A.; Mathew,, Y. (2001), "Synthesis of a C1-C21 Subunit of the Protein Phosphatase Inhibitor Tautomycin: A Formal Total Synthesis", J. Org. Chem.: 2373–1379, doi:10.1021/jo0056951
- 1 2 Tamao, K.; Ishida, N.; Kumada, M.; Henning, R.; Plaut, H.E. (1983), "Diisopropoxymethylsilyl)methyl Grignard reagent: a new, practically useful nucleophilic hydroxymethylating agent", J. Org. Chem.: 2120–2122, doi:10.1021/jo00160a046
- 1 2 Tamao, K.; Ishida, N.; Tanaka, T.; Kumada, M. (1983), "Silafunctional compounds in organic synthesis. Part 20. Hydrogen peroxide oxidation of the silicon-carbon bond in organoalkoxysilanes", Organometallics, 2: 1694–1696, doi:10.1021/om50005a041
- 1 2 "Name Reactions: A Collection of Detailed Reaction Mechanisms," 2nd ed. by Li, J. J. (2003), 404, CODEN: ADSDEO; ISBN 3-540-40203-9
- ↑ "Frontiers of Organosilicon Chemistry," by Tamao, K.; Hayashi, T.; Ito, Y. (1991), 197–207.
- ↑ Fleming, I.; Sanderson, P.E.J. (1987), "A one-pot conversion of the phenyldimethylsilyl group into a hydroxyl group", Tetrahedron Lett., 52: 4229–4232, doi:10.1016/s0040-4039(00)95587-4
- 1 2 Sunderhaus, J D.; Lam, H.; Dudley, G B. (2003), "Oxidation of Carbon−Silicon Bonds: The Dramatic Advantage of Strained Siletanes", Org. Lett.: 2120–2122, doi:10.1021/ol035695y
- 1 2 3 Tamao, K.; Kumada, M. (1984), "Silafunctional Compounds in organic Synthesis. 21.1 Hydrogen peroxide Oxidation of alkenyl (alkoxy)silanes", Tetrahedron Lett.: 321–324, doi:10.1016/S0040-4039(00)99873-3
- 1 2 Barrett, Anthony G. M.; Head, John; Smith, Marie L.; Stock, Nicholas S.; White, A. J. P.; Williams, D. J. (1999). "Fleming−Tamao Oxidation and Masked Hydroxyl Functionality: Total Synthesis of (+)-Pramanicin and Structural Elucidation of the Antifungal Natural Product (−)-Pramanicin". The Journal of Organic Chemistry. 64 (16): 6005–6018. doi:10.1021/jo9905672. ISSN 0022-3263.
- ↑ Woerpel, K.A.; Tenenbaum, J.M. (2003), "Use of an Aminosilyllithium for the Diastereoselective Synthesis of Diphenyl Oxasilacyclopentane Acetals and Polyols", Org. Lett.: 4325–4327, doi:10.1021/ol035577a
- ↑ "Abstracts of Papers, 222nd ACS Meeting, Chicago" by Hara K.; Moralee, A.C.; Ojima, I. (2001), ORGN-089.