Electrophilic amination
Electrophilic amination is a chemical process involving the formation of a carbon–nitrogen bond through the reaction of a nucleophilic carbanion with an electrophilic source of nitrogen.[1]
Introduction
Electrophilic amination reactions can be classified as either additions or substitutions. Although the resulting product is not always an amine, these reactions are unified by the formation of a carbon–nitrogen bond and the use of an electrophilic aminating agent. A wide variety of electrophiles have been used; for substitutions, these are most commonly amines substituted with electron-withdrawing groups: chloramines, hydroxylamines, hydrazines, and oxaziridines, for instance. Addition reactions have employed imines, oximes, azides, azo compounds, and others.

Mechanism and stereochemistry
Prevailing mechanisms
A nitrogen bound to both a good electrofuge and a good nucleofuge is known as a nitrenoid (for its resemblance to a nitrene).[2] Nitrenes lack a full octet of electrons are thus highly electrophilic; nitrenoids exhibit analogous behavior and are often good substrates for electrophilic amination reactions. Nitrenoids can be generated from O-alkylhydroxylamines containing an N-H bond via deprotonation or from O-alkyloximes via nucleophilic addition. These intermediates react with carbanions to give substituted amines. Other electron-deficient, sp3 amination reagents react by similar mechanisms to give substitution products.[3]
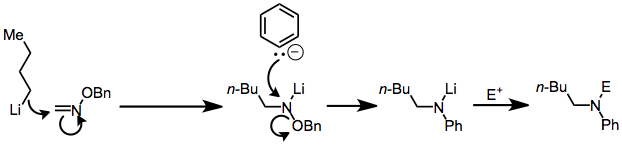
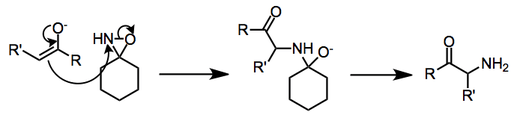
In aminations involving oxaziridines, nucleophilic attack takes place on the nitrogen atom of the three-membered ring. For some substrates (α-cyano ketones, for example), the resulting alkoxide reacts further to afford unexpected products. Straightforward β elimination of the alkoxide leads to the formation of an amine.[4]
Additions across pi bonds appear to proceed by typical nucleophilic addition pathways in most cases. Alkyl-, aryl-, and heteroaryllithium reagents add to azides to afford triazene salts.[5] Reduction of these salts leads to amines, although they also may be converted to azides upon acidic workup with overall elimination of sulfinic acid.

Enantioselective variants
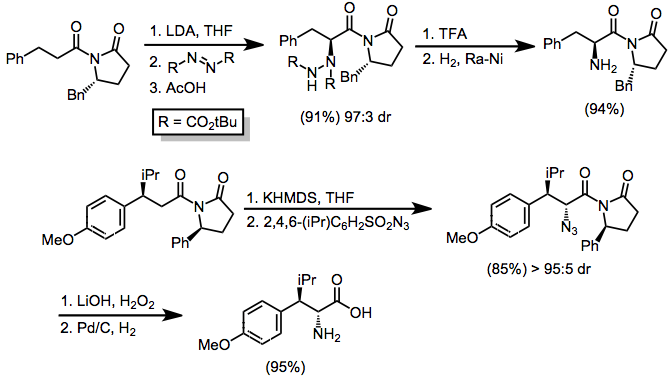
The most synthetically useful aminations of enolate anions employ N-acyloxazolidinone substrates.[6] The chiral auxiliaries on these compounds are easily removed after hydrazine formation (with azo compounds) or azidation (with trisyl azide). Azidation using the latter reagent is more efficient than bromination followed by nucleophilic substitution by the azide anion[7] Palladium on carbon and hydrogen gas reduce both azide and hydrazide products (the latter only after conversion to the hydrazine).
Scope and limitations
Aminating reagents
Electrophilic aminating reagents rely on the presence of an electron-withdrawing functional group attached to nitrogen. A variety of hydroxylamine derivatives have been used for this purpose. Sulfonylhydroxylamines are able to aminate a wide array of carbanions.[8]

Azo compounds afford hydrazines after addition to the N=N bond. These additions have been rendered enantioselective through the use of chiral auxiliaries (see above) and chiral catalysts.[9] Although the enantioselectivity of the proline-catalyzed process is good, yields are low and reaction times are long.

Upon treatment with sulfonyl azides, a variety of Grignard reagents or enolates may be converted into azides or amines.[10] A significant side reaction that occurs under these conditions is the diazo transfer reaction: instead of fragmenting into an azide and sulfinic acid, the intermediate triazene salt may break down to a diazo compound and sulfonamide. Changing workup conditions may favor one product over another. In general, for reactions of enolates substituted with Evans oxazolidinones, trifluoroacetic acid promotes diazo transfer while acetic acid encourages azidation (the reasons why are unclear).[11] Solvent and the enolate counterion also influence the observed ratio of diazo to azide products.[12]

Other electrophilic aminating reagents include oxaziridines,[13] diazo compounds,[14] and in rare cases, imines.[15]
Organometallic substrates
The scope of organometallic reagents that may be aminated by electrophilic methods is large. Alkyl Grignard reagents,[16] alkylithium compounds,[17] alkylzinc compounds,[18] and alkylcuprates[19] have been aminated with electrophilic reagents successfully. Among sp2-centered carbanions, vinyllithium compounds,[8] vinylcuprates,[20] and vinyl Grignard reagents[21] react with electrophilic aminating reagents to afford enamines.
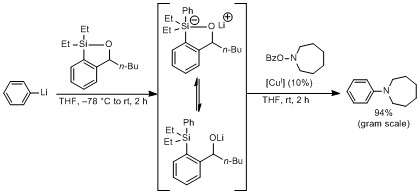
Aryl and heteroaryl organolithium reagents undergo efficient electrophilic amination under copper (I) catalyzed condition mediated by recoverable silicon reagents, termed siloxane transfer agents.[22]
The scope of sp-centered carbanions is limited to alkynylcuprates.[23] Enolates and silyl enol ethers, the most widely used class of carbon nucleophiles in electrophilic amination reactions, participate in amination,[24] adization[25] and hydrazination[7] reactions.
The primary application of alkylmetal reagents in electrophilic amination reactions is the synthesis of hindered amines, many of which are difficult to prepare through nucleophilic displacement with an alkyl halide (nucleophilic amination). For instance, in the presence of a copper(II) catalyst, bulky organozinc reagents react with O-acylhydroxylamines to afford hindered amines.

Allylic metal species can be used to prepare allylic amines through electrophilic amination. Although allylic amines are usually prepared through nucleophilic amination of allylic halides, a few examples of electrophlic amination of allylic substrates are known. In the example below, an allylic zirconium reagent (obtained by hydrozirconation) is trapped with an O-alkylhydroxylamine.[26]

The electrophilic amination of enolates yields α-amino carbonyl compounds. When chiral oxazolidinones are used in conjunction with azo compounds, enantioselectivity is observed (see above). BINAP can also be used for this purpose in the amination of silyl enol ethers.

Aryl and heteroaryl organometallic reagents undergo many of the same transformations as their aliphatic counterparts. Formation of amines, hydrazines, and azides is possible through the use of various electrophilic aminating reagents. An example employing a nitrenoid reagent is shown below.[27]
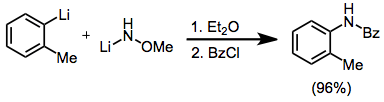
Intramolecular amination is possible, and has been used to prepare small and medium rings. In the example below, deprotonation of an activated methylene compound containing an O-phosphinoylhydroxylamine led to the cyclic amine shown.[28]
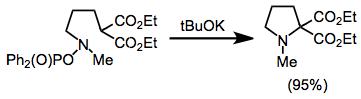
Comparison with other methods
Several other methods for the electrophilic formation of C-N bonds are available. Nitrites and nitrates can be used to form oximes and nitro compounds, respectively. Additionally, organoboranes can serve the role of the nucleophile and often provide higher yields with fewer complications than analogous carbanions. The Neber rearrangement offers an alternative to electrophilic amination through the intermediacy of an azirine.

Typical conditions
The wide variety of electrophilic aminating reagents precludes generalization of reaction conditions. Electrophilic nitrogen sources are, however, either toxic or explosive in general. Great care should be taken while handling these reagents. Many electrophilic nitrogen sources do not provide amines immediately, but a number of methods exist to generate the corresponding amines.
- Tosylamines: tributyltin hydride
- Azo compounds: H2/Pd
- Triazenes: sodium borohydride
- Azides: H2/Pd, H2/Pt, lithium aluminum hydride, triphenylphosphine
Conversion to other nitrogen-containing functionality, including enamines,[29] imines,[30] and amides,[31] is also possible.
References
- ↑ Ciganek, E. Org. React. 2008, 72, 1–86. doi:10.1002/0471264180.or072.01
- ↑ Starkov, P., Jamison, T. J., Marek. I. M. Chem. Eur. J. 2015, 21, doi:10.1002/chem.201405779
- ↑ Beak, P.; Selling, G. W. J. Org. Chem. 1989, 54, 5574.
- ↑ Page, P. C. B.; Limousin, C.; Murrell, V. L. J. Org. Chem. 2002, 67, 7787
- ↑ Reed, J. N.; Snieckus, V. Tetrahedron Lett. 1984, 25, 5505.
- ↑ Evans, D. A.; Britton, T. C. J. Am. Chem. Soc. 1987, 109, 6881.
- 1 2 Evans, D. A.; Britton, T. C.; Dorow, R. L.; Dellaria, J. F., Jr. Tetrahedron 1988, 44, 5525.
- 1 2 Boche, G.; Mayer, N.; Bernheim, M.; Wagner, K. Angew. Chem. Int. Ed. Engl. 1978, 17, 687.
- ↑ Baumann, T.; Vogt, H.; Bräse, S. Eur. J. Org. Chem. 2007, 266.
- ↑ Smith, P. A. S.; Rowe, C. D.; Bruner, L. B. J. Org. Chem. 1969, 34, 3430.
- ↑ Shiro, Y.; Kato, K.; Fujii, M.; Ida, Y.; Akita, H. Tetrahedron 2006, 62, 8687.
- ↑ Evans, D. A.; Britton, T. C.; Ellman, J. A. Dorow, R. L. J. Am. Chem. Soc. 1990, 112, 4011.
- ↑ Andreae, S.; Schmitz, E. Synthesis 1991, 327.
- ↑ Enders, E. In Methoden der organischen Chemie (Houben-Weyl); Georg Thieme Verlag: Stuttgart, 1967; Vol. 10/2, p 456.
- ↑ Niwa, Y.; Takayama, K.; Shimizu, M. Bull. Chem. Soc. Jpn. 2002, 75, 1819.
- ↑ Kitamura, M.; Suga, T.; Chiba, S.; Narasaka, K. Org. Lett. 2004, 6, 4619.
- ↑ Kraus, G. A. U. S. Patent 5,599,998 (1997).
- ↑ Sinha, P.; Kofink, C. C.; Knochel, P. Org. Lett. 2006. 8, 3741.
- ↑ Alberti, A.; Canè, F.; Dembech, P.; Lazzari. D.; Ricci, A.; Seconi, G. J. Org. Chem. 1996, 61, 1677
- ↑ Cané, F.; Brancaleoni, D.; Dembech, P.; Ricci, A.; Seconi, G. Synthesis 1997, 545.
- ↑ Akimova, G. S.; Kolokol'tseva, I. G.; Chistokletov, V. N.; Petrov, A. A. Zh. Org. Khim. 1968, 4, 954; Engl. Transl. p 927.
- ↑ Nguyen, M. H.; Smith A. B. III. Org. Lett 2013, 4872
- ↑ Boche, G.; Bernheim, M.; Niessner, M. Angew. Chem. Int. Ed. Engl. 1983, 22, 53.
- ↑ Evans, D. A.; Faul, M. M.; Bilodeau, M. T. J. Am. Chem. Soc. 1994, 116, 2742.
- ↑ Shimizu, M.; Nemoto, H.; Kakuda, H.; Takahata, H. Heterocycles 2003, 59, 245.
- ↑ Berman, A. M.; Johnson, J. S. J. Org. Chem. 2006, 71, 219.
- ↑ Beak, P.; Basha, A.; Kokko, B.: Loo, D. K. J. Am. Chem. Soc. 1986, 108, 6016.
- ↑ Sheradsky, T.; Yusupova, L. Tetrahedron Lett. 1995, 36, 7701.
- ↑ Magnus, P.; Barth, L. Tetrahedron 1995, 51, 11075.
- ↑ Magnus, P.; Mugrage, B. J. Am. Chem. Soc. 1990, 112, 462.
- ↑ Marshalkin, M. F.; Yakhontov, L. N. Uspekhi Khim. 1986, 55, 1785