Disseminated intravascular coagulation
| Disseminated intravascular coagulation or Disseminated intravascular coagulopathy | |
|---|---|
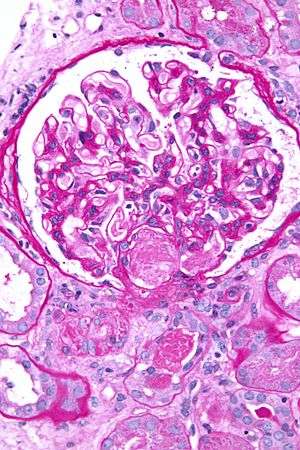 | |
| Micrograph showing an acute thrombotic microangiopathy, the histologic correlate of DIC, in a kidney biopsy. A thrombus is present in the hilum of the glomerulus (center of image). PAS stain. | |
| Classification and external resources | |
| Specialty | Haematology |
| ICD-10 | D65 |
| ICD-9-CM | 286.6 |
| DiseasesDB | 3765 |
| MedlinePlus | 000573 |
| eMedicine | med/577 emerg/150 |
| MeSH | D004211 |
Disseminated intravascular coagulation (DIC), also known as disseminated intravascular coagulopathy or less commonly as consumptive coagulopathy, is a pathological process characterized by the widespread activation of the clotting cascade that results in the formation of blood clots in the small blood vessels throughout the body. This leads to compromise of tissue blood flow and can ultimately lead to multiple organ damage. In addition, as the coagulation process consumes clotting factors and platelets, normal clotting is disrupted and severe bleeding can occur from various sites.
DIC does not occur by itself but only as a complicating factor from another underlying condition, usually in those with a critical illness. The combination of widespread loss of tissue blood flow and simultaneous bleeding leads to an increased risk of death in addition to that posed by the underlying disease. DIC can be overt and severe in some cases, but milder and insidious in others. The diagnosis of DIC depends on the findings of characteristic laboratory tests and clinical background. Treatment is mainly geared towards the underlying condition.[1][2][3][4][5]
Signs and symptoms
In DIC, the underlying cause usually leads to symptoms and signs, and DIC is discovered on laboratory testing. The onset of DIC can be sudden, as in endotoxic shock or amniotic fluid embolism, or it may be insidious and chronic, as in cancer. DIC can lead to multiorgan failure and widespread bleeding.[5]
Causes
DIC can occur in the following conditions:[3][5][6]
- Solid tumors and blood cancers (particularly acute promyelocytic leukemia)
- Obstetric complications: abruptio placentae, pre-eclampsia or eclampsia, amniotic fluid embolism, retained intrauterine fetal demise, septic abortion ,post partum haemorrhage
- Massive tissue injury: severe trauma, burns, hyperthermia, rhabdomyolysis, extensive surgery
- Sepsis or severe infection of any kind (infections by nearly all microorganisms can cause DIC, though bacterial infections are the most common): bacterial (Gram-negative and Gram-positive sepsis), viral, fungal, or protozoan infections
- Transfusion reactions (i.e., ABO incompatibility haemolytic reactions)
- Severe allergic or toxic reactions (i.e. snake or viper venom)
- Giant haemangiomas (Kasabach-Merritt syndrome)
- Large aortic aneurysms
Liver disease, HELLP syndrome, thrombotic thrombocytopenic purpura/Haemolytic uremic syndrome, and malignant hypertension may mimic DIC but do not occur via the same pathways.
Pathophysiology
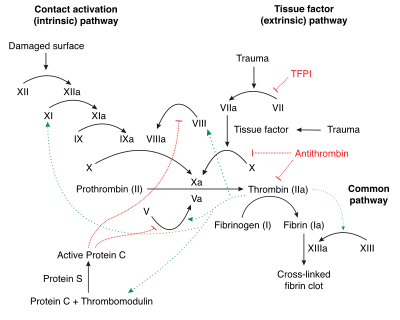
Under homeostatic conditions, the body is maintained in a finely tuned balance of coagulation and fibrinolysis. The activation of the coagulation cascade yields thrombin that converts fibrinogen to fibrin; the stable fibrin clot being the final product of haemostasis. The fibrinolytic system then functions to break down fibrinogen and fibrin. Activation of the fibrinolytic system generates plasmin (in the presence of thrombin), which is responsible for the lysis of fibrin clots. The breakdown of fibrinogen and fibrin results in polypeptides called fibrin degradation products (FDPs) or fibrin split products (FSPs). In a state of homeostasis, the presence of plasmin is critical, as it is the central proteolytic enzyme of coagulation and is also necessary for the breakdown of clots, or fibrinolysis.
In DIC, the processes of coagulation and fibrinolysis are dysregulated, and the result is widespread clotting with resultant bleeding. Regardless of the triggering event of DIC, once initiated, the pathophysiology of DIC is similar in all conditions. One critical mediator of DIC is the release of a transmembrane glycoprotein called tissue factor (TF). TF is present on the surface of many cell types (including endothelial cells, macrophages, and monocytes) and is not normally in contact with the general circulation, but is exposed to the circulation after vascular damage. For example, TF is released in response to exposure to cytokines (particularly interleukin 1), tumor necrosis factor, and endotoxin.[7] This plays a major role in the development of DIC in septic conditions. TF is also abundant in tissues of the lungs, brain, and placenta. This helps to explain why DIC readily develops in patients with extensive trauma. Upon exposure to blood and platelets, TF binds with activated factor VIIa (normally present in trace amounts in the blood), forming the extrinsic tenase complex. This complex further activates factor IX and X to IXa and Xa, respectively, leading to the common coagulation pathway and the subsequent formation of thrombin and fibrin.[4]
The release of endotoxin is the mechanism by which Gram-negative sepsis provokes DIC. In acute promyelocytic leukemia, treatment causes the destruction of leukemic granulocyte precursors, resulting in the release of large amounts of proteolytic enzymes from their storage granules, causing microvascular damage. Other malignancies may enhance the expression of various oncogenes that result in the release of TF and plasminogen activator inhibitor-1 (PAI-1), which prevents fibrinolysis.[8]
Excess circulating thrombin results from the excess activation of the coagulation cascade. The excess thrombin cleaves fibrinogen, which ultimately leaves behind multiple fibrin clots in the circulation. These excess clots trap platelets to become larger clots, which leads to microvascular and macrovascular thrombosis. This lodging of clots in the microcirculation, in the large vessels, and in the organs is what leads to the ischemia, impaired organ perfusion, and end-organ damage that occurs with DIC.
Coagulation inhibitors are also consumed in this process. Decreased inhibitor levels will permit more clotting so that a positive feedback loop develops in which increased clotting leads to more clotting. At the same time, thrombocytopenia occurs and this has been attributed to the entrapment and consumption of platelets. Clotting factors are consumed in the development of multiple clots, which contributes to the bleeding seen with DIC.
Simultaneously, excess circulating thrombin assists in the conversion of plasminogen to plasmin, resulting in fibrinolysis. The breakdown of clots results in an excess of FDPs, which have powerful anticoagulant properties, contributing to hemorrhage. The excess plasmin also activates the complement and kinin systems. Activation of these systems leads to many of the clinical symptoms that patients experiencing DIC exhibit, such as shock, hypotension, and increased vascular permeability. The acute form of DIC is considered an extreme expression of the intravascular coagulation process with a complete breakdown of the normal homeostatic boundaries. DIC is associated with a poor prognosis and a high mortality rate.
There has been a recent challenge however to the basic assumptions and interpretations of the pathophysiology of DIC. A study of sepsis and DIC in animal models has shown that a highly expressed receptor on the surface of hepatocytes, termed the Ashwell-Morell receptor, is responsible for thrombocytopenia in bacteremia and sepsis due to Streptococcus pneumoniae (SPN) and possibly other pathogens. The thrombocytopenia observed in SPN sepsis was not due to increased consumption of coagulation factors such as platelets, but instead was the result of this receptor's activity enabling hepatocytes to ingest and rapidly clear platelets from circulation.[9] By removing pro-thrombotic components before they participate in the coagulopathy of DIC, the Ashwell-Morell receptor lessens the severity of DIC, reducing thrombosis and tissue necrosis, and promoting survival. The hemorrhage observed in DIC and among some tissues lacking this receptor may thereby be secondary to increased thrombosis with loss of the mechanical vascular barrier. This discovery has possible significant clinical implications in devising new approaches to reducing the morbidity and mortality of DIC.
Diagnosis
The diagnosis of DIC is not made on a single laboratory value, but rather the constellation of laboratory markers and a consistent history of an illness known to cause DIC. Laboratory markers consistent with DIC include:[1][4][10]
- Characteristic history (this is important because severe liver disease can essentially have the same laboratory findings as DIC)
- Prolongation of the prothrombin time (PT) and the activated partial thromboplastin time (aPTT) reflect the underlying consumption and impaired synthesis of the coagulation cascade.
- Fibrinogen level has initially thought to be useful in the diagnosis of DIC but because it is an acute phase reactant, it will be elevated due to the underlying inflammatory condition. Therefore, a normal (or even elevated) level can occur in over 57% of cases. A low level, however, is more consistent with the consumptive process of DIC.
- A rapidly declining platelet count
- High levels of fibrin degradation products, including D-dimer, are found owing to the intense fibrinolytic activity stimulated by the presence of fibrin in the circulation.
- The peripheral blood smear may show fragmented red blood cells (known as schistocytes) due to shear stress from thrombi. However, this finding is neither sensitive nor specific for DIC
A diagnostic algorithm has been proposed by the International Society of Thrombosis and Haemostasis. This algorithm appears to be 91% sensitive and 97% specific for the diagnosis of overt DIC. A score of 5 or higher is compatible with DIC and it is recommended that the score is repeated daily, while a score below 5 is suggestive but not affirmative for DIC and it is recommended that it is repeated only occasionally:[10][11] It has been recommended that a scoring system be used in the diagnosis and management of DIC in terms of improving outcome.[12]
- Presence of an underlying disorder known to be associated with DIC (no=0, yes=2)
- Global coagulation results
- Platelet count (>100k = 0, <100 = 1, <50 = 2)
- Fibrin degradation products such as D-Dimer (no increase = 0, moderate increase = 2, strong increase = 3)
- Prolonged prothrombin time (<3 sec = 0, >3 sec = 1, >6 sec = 2)
- Fibrinogen level (> 1.0g/L = 0; < 1.0g/L = 1[13])
Treatment
Treatment of DIC is centered around treating the underlying condition. Transfusions of platelets or fresh frozen plasma can be considered in cases of significant bleeding, or those with a planned invasive procedure. The target goal of such transfusion depends on the clinical situation. Cryoprecipitate can be considered in those with a low fibrinogen level.
Treatment of thrombosis with anticoagulants such as heparin is rarely used due to the risk of bleeding.
Recombinant human activated protein C was previously recommended in those with severe sepsis and DIC, but drotrecogin alfa has been shown to confer no benefit and was withdrawn from the market in 2011.
Recombinant factor VII has been proposed as a "last resort" in those with severe hemorrhage due to obstetric or other causes, but conclusions about its use are still insufficient.[14]
Prognosis
Prognosis varies depending on the underlying disorder, and the extent of the intravascular thrombosis (clotting). The prognosis for those with DIC, regardless of cause, is often grim: Between 10% and 50% of patients will die.[15] DIC with sepsis (infection) has a significantly higher rate of death than DIC associated with trauma.[15]
Epidemiology
DIC is observed in approximately 1% of academic hospital admissions.[16] DIC occurs at higher rates in patients with bacterial sepsis (83%),[17] severe trauma (31%),[18] and cancer (6.8%).[19]
See also
- Sepsis
- Septic shock
- Blood clotting tests
- DIC is discussed in Michael Crichton's 1969 technothriller The Andromeda Strain, and its movie adaptations.
References
- 1 2 Levi, M (2007). "Disseminated Intravascular Coagulation". Critical Care Medicine. 35 (9): 2191–2195. doi:10.1097/01.CCM.0000281468.94108.4B. PMID 17855836.
- ↑ Levi, M; ten Cate, H (1999). "Disseminated Intravascular Coagulation". New England Journal of Medicine. 341 (8): 586–592. doi:10.1056/NEJM199908193410807. PMID 10451465.
- 1 2 Davidson's Principles and Practice of Medicine (19 ed.). Churchill Livingstone. 2002. ISBN 0-443-07036-9.
- 1 2 3 Haematology: Basic Principles and Practice (6 ed.). Elsevier Saunders. 2012. ISBN 1437729282.
- 1 2 3 Robbins, Stanley L.; Cotran, Ramzi S.; Kumar, Vinay; Collins, Tucker (1999). Robbins' Pathologic Basis of Disease (6 ed.). Philadelphia: Saunders. ISBN 0-7216-7335-X.
- ↑ Clark, Michael; Kumar, Parveen J. (1998). Clinical Medicine: A Textbook for Medical Students and Doctors (4 ed.). Philadelphia: W.B. Saunders. ISBN 0-7020-2458-9.
- ↑ Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; & Mitchell, Richard N. (2007). Robbins Basic Pathology (8th ed.). Saunders Elsevier. pp. 469-471 ISBN 978-1-4160-2973-1
- ↑ Rak J, Yu JL, Luyendyk J, Mackman N (2006). "Oncogenes, trousseau syndrome, and cancer-related changes in the coagulome of mice and humans". Cancer Res. 66 (22): 10643–6. doi:10.1158/0008-5472.CAN-06-2350. PMID 17108099.
- ↑ Grewal, PK; Uchiyama, S; Ditto, D; Varki, N; Le, DT; Nizet, V; Marth, JD (June 2008). "The Ashwell receptor mitigates the lethal coagulopathy of sepsis.". Nature Medicine. 14 (6): 648–55. doi:10.1038/nm1760. PMC 2853759
 . PMID 18488037.
. PMID 18488037. - 1 2 Levi, M; Toh, C-H; et al. (2009). "Guidelines for the diagnosis and management of disseminated intravascular coagulation". British Journal of Haematology. 145 (5): 24–33. doi:10.1111/j.1365-2141.2009.07600.x. PMID 19222477.
- ↑ Taylor, F; Toh, C-h; et al. (2001). "Towards Definition, Clinical and Laboratory Criteria, and a Scoring System for Disseminated Intravascular Coagulation". Thrombosis and Haemostasis. 86 (5): 1327–30. PMID 11816725.
- ↑ Gando, S (2012). "The Utility of a Diagnostic Scoring System for Disseminated Intravascular Coagulation". Critical Care Clinics. 28 (3): 378–88. doi:10.1016/j.ccc.2012.04.004. PMID 22713612.
- ↑ http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2009.07600.x/full
- ↑ Franchini, M; Manzato, F; Salvagno GL; et al. (2007). "Potential role of recombinant activated factor VII for the treatment of severe bleeding associated with disseminated intravascular coagulation: a systematic review". Blood Coagul Fibrinolysis. 18 (7): 589–93. doi:10.1097/MBC.0b013e32822d2a3c. PMID 17890943.
- 1 2 Becker, Joseph U and Charles R Wira. Disseminated intravascular coagulation at eMedicine, 10 September 2009
- ↑ Matsuda, T (Jan–Feb 1996). "Clinical aspects of DIC--disseminated intravascular coagulation". Pol J Pharmacol. 48 (1): 73–5. PMID 9112631.
- ↑ Smith, OP (1997). "Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans.". Lancet. 350 (9091): 1590. doi:10.1016/s0140-6736(97)06356-3.
- ↑ Gando, S (1999). "Disseminated intravascular coagulation and sustained systemic inflammatory response syndrome predict organ dysfunctions after trauma: application of clinical decision analysis.". Ann Surg. 229 (1): 121. doi:10.1097/00000658-199901000-00016.
- ↑ Sallah, S (2001). "Disseminated intravascular coagulation in solid tumors: clinical and pathologic study.". Thromb Haemost. 86 (3): 828.