Acute lymphoblastic leukemia
| Acute lymphoblastic leukemia | |
|---|---|
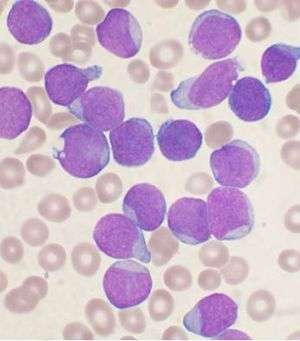 | |
| Classification and external resources | |
| Specialty | Hematology and oncology |
| ICD-10 | C91.0 |
| ICD-9-CM | 204.0 |
| ICD-O | M9835/3 |
| DiseasesDB | 195 |
| eMedicine | med/3146 ped/2587 |
| MeSH | D054198 |
Acute lymphoblastic leukemia, also known as acute lymphocytic leukemia or acute lymphoid leukemia (ALL), is an acute form of leukemia, or cancer of the white blood cells, characterized by the overproduction and accumulation of cancerous, immature white blood cells, known as lymphoblasts.[1] In persons with ALL, lymphoblasts are overproduced in the bone marrow and continuously multiply, causing damage and death by inhibiting the production of normal cells (such as red and white blood cells and platelets) in the bone marrow and by spreading (infiltrating) to other organs. ALL is most common in childhood, with a peak incidence at 2–5 years of age and another peak in old age.[1]
The symptoms of ALL are indicative of a reduced production of functional blood cells, because leukemia wastes the resources of the bone marrow that are normally used to produce new, functioning blood cells.[1] These symptoms can include fever, increased risk of infection (especially bacterial infections like pneumonia, due to neutropenia; symptoms of such an infection include shortness of breath, chest pain, cough, vomiting, changes in bowel or bladder habits), increased tendency to bleed (due to thrombocytopenia), and signs indicative of anemia, including pallor, tachycardia (high heart rate), fatigue, and headache.[1]
About 6,000 cases are reported in the United States every year.[2] Internationally, ALL is more common in Caucasians than in Africans; it is more common in Hispanics and in Latin America.[3]:1617[4] Cure is a realistic goal and is achieved in more than 80% of affected children, although only 20-40% of adults are cured.[1] "Acute" is defined by the World Health organization standards, in which greater than 20% of the cells in the bone marrow are blasts. Chronic lymphocytic leukemia is defined as having less than 20% blasts in the bone marrow.
ALL was one of the first cancers for which an effective chemotherapeutic treatment was developed. Antifolates like aminopterin and methotrexate were developed in the late 1940s by Sidney Farber and Yellapragada Subbarow.[5][6] At that time, a doctor did not need a patient's or parent's consent to try an experimental treatment as the Nuremberg code had not yet been signed. Desperate to save his patients, Farber initially tried folic acid supplementation as a treatment for ALL. This had disastrous consequences and he likely accelerated the children's deaths.[6]
Signs and symptoms

Initial symptoms are not specific to ALL, but worsen to the point that medical help is sought. They result from the lack of normal and healthy blood cells because they are crowded out by malignant and immature leukocytes (white blood cells). Therefore, people with ALL experience symptoms from malfunctioning of their erythrocytes (red blood cells), leukocytes, and platelets. Laboratory tests that might show abnormalities include blood count tests, renal function tests, electrolyte tests, and liver enzyme tests.[1]
The signs and symptoms of ALL are variable but follow from bone marrow replacement and/or organ infiltration.[1]
- Generalized weakness and fatigue
- Anemia
- Dizziness
- Frequent or unexplained fever and infection
- Weight loss and/or loss of appetite
- Excessive and unexplained bruising
- Bone pain, joint pain (caused by the spread of "blast" cells to the surface of the bone or into the joint from the marrow cavity)
- Breathlessness
- Enlarged lymph nodes, liver and/or spleen
- Pitting edema (swelling) in the lower limbs and/or abdomen
- Petechiae, which are tiny red spots or lines in the skin due to low platelet levels
Pathophysiology

In general, cancer is caused by damage to DNA that leads to uncontrolled cellular growth and spreads throughout the body, either by increasing chemical signals that cause growth or by interrupting chemical signals that control growth. Damage can be caused through the formation of fusion genes, as well as the dysregulation of a proto-oncogene via juxtaposition of it to the promoter of another gene, e.g. the T-cell receptor gene. This damage may be caused by environmental factors such as chemicals, drugs or radiation, and occurs naturally during mitosis or other normal processes (although cells have numerous mechanisms of DNA repair that help to reduce this).
ALL is associated with exposure to radiation and chemicals in animals and humans. High level radiation exposure is a known risk factor for developing leukemia, as found by studies of survivors of atom bomb exposure in Hiroshima and Nagasaki.[7] In animals, exposure to benzene and other chemicals can cause leukemia. Epidemiological studies have associated leukemia with workplace exposure to chemicals, but these studies are not as conclusive. Some evidence suggests that secondary leukemia can develop in individuals treated for other cancers with radiation and chemotherapy as a result of that treatment.[8]
Diagnosis
Diagnosing ALL begins with a medical history, physical examination, complete blood count, and blood smears. Because the symptoms are so general, many other diseases with similar symptoms must be excluded. Typically, the higher the white blood cell count, the worse the prognosis.[9] Blast cells are seen on blood smear in the majority of cases (blast cells are precursors — stem cells — to all immune cell lines). A bone marrow biopsy provides conclusive proof of ALL.[10] A lumbar puncture (also known as a spinal tap) will indicate whether the spinal column and brain have been invaded.
Pathological examination, cytogenetics (in particular the presence of Philadelphia chromosome), and immunophenotyping establish whether myeloblastic (neutrophils, eosinophils, or basophils) or lymphoblastic (B lymphocytes or T lymphocytes) cells are the problem. RNA testing can establish how aggressive the disease is; different mutations have been associated with shorter or longer survival. Immunohistochemicaltesting may reveal TdT or CALLA antigens on the surface of leukemic cells. TdT is a protein expressed early in the development of pre-T and pre-B cells, whereas CALLA is an antigen found in 80% of ALL cases and also in the "blast crisis" of CML.
Medical imaging (such as ultrasound or CT scanning) can find invasion of other organs commonly the lung, liver, spleen, lymph nodes, brain, kidneys, and reproductive organs.[11]
-
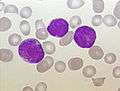
acute lymphoblastic leukemia (ALL), peripheral blood of a child, Pappenheim stain, magnification x100
-

bone marrow smear (large magnification) from a patient with acute lymphoblastic leukemia
-

bone marrow smear from a patient with acute lymphoblastic leukemia
Cytogenetics
Cytogenetic translocations associated with specific molecular genetic abnormalities in ALL
| Cytogenetic translocation | Molecular genetic abnormality | % |
|---|---|---|
| cryptic t(12;21) | TEL-AML1 fusion[12] | 25.4%[13] |
| t(1;19)(q23;p13) | E2A-PBX (PBX1) fusion[14] | 4.8%[13] |
| t(9;22)(q34;q11) | BCR-ABL fusion(P185)[15] | 1.6%[13] |
| t(4;11)(q21;q23) | MLL-AF4 fusion[16] | 1.6%[13] |
| t(8;14)(q24;q32) | IGH-MYC fusion[17] | |
| t(11;14)(p13;q11) | TCR-RBTN2 fusion[18] |
12;21 is the most common translocation and portends a good prognosis. 4;11 is the most common in children under 12 months and portends a poor prognosis.
Classification
As ALL is not a solid tumor, the TNM notation, used in solid cancers, has no relevancy. Lymphoblastic lymphoma is a rare type of non-Hodgkin lymphoma, a result of abnormal adaptive immune cells, typically T-cells. It usually occurs in children.
Prior to 2008, subtyping of all acute leukemias (including acute myelogenous leukemia, AML) used the French-American-British (FAB) classification,[19] in which ALL was classified as:
- ALL-L1: small uniform cells
- ALL-L2: large varied cells
- ALL-L3: large varied cells with vacuoles (bubble-like features).
The FAB scheme had only a limited impact on treatment choice.[20]:491 In 2008, the World Health Organization scheme identified three therapeutically distinct categories. These are identified by immunophenotyping of surface markers of the abnormal lymphocytes:[20]:492[21]:1529
- B-lymphoblastic ALL (this category can be subdivided according to the correlation of the ALL cell immunophenotype with the stages of normal B-cell development)
- Burkitt ALL (corresponds to ALL-L3)
- T-cell ALL.
This subtyping helps determine the prognosis and the most appropriate treatment in treating ALL. It is substantially amplified by cytogenetics and molecular diagnostics tests.[21]:1531–1535
- 1. Acute lymphoblastic leukemia/lymphoma. Synonyms: Former FAB L1/L2
- i. Precursor B acute lymphoblastic leukemia/lymphoma. Cytogenetic subtypes:[22][23]
- t(12;21)(p12,q22) TEL/AML-1
- t(1;19)(q23;p13) PBX/E2A
- t(9;22)(q34;q11) ABL/BCR
- T(V,11)(V;q23) V/MLL
- ii. Precursor T acute lymphoblastic leukemia/lymphoma
- i. Precursor B acute lymphoblastic leukemia/lymphoma. Cytogenetic subtypes:[22][23]
- 2. Burkitt's leukemia/lymphoma. Synonyms: Former FAB L3
- 3. Biphenotypic acute leukemia
Variant features
- Acute lymphoblastic leukemia with cytoplasmic granules
- Aplastic presentation of ALL
- Acute lymphoblastic leukemia with eosinophilia
- Relapse of lymphoblastic leukemia
- Secondary ALL
Immunophenotyping
The use of a TdT assay and a panel of monoclonal antibodies (MoAbs) to T cell and B cell associated antigens will identify almost all cases of ALL.
Immunophenotypic categories of acute lymphoblastic leukemia (ALL):
| Types | FAB Class | Tdt | T cell associate antigen | B cell associate antigen | c Ig | s Ig |
|---|---|---|---|---|---|---|
| Precursor B | L1,L2 | + | - | + | -/+ | - |
| Precursor T | L1,L2 | + | + | - | - | - |
| B-cell | L3 | - | - | + | - | + |
Treatment
The earlier ALL is detected, the more effective the treatment. The aim is to induce a lasting remission, defined as the absence of detectable cancer cells in the body (usually less than 5% blast cells in the bone marrow).
Treatment for acute leukemia can include chemotherapy, steroids, radiation therapy, intensive combined treatments (including bone marrow or stem cell transplants), and growth factors.[24]
Chemotherapy
Chemotherapy is the initial treatment of choice. Most ALL patients will receive a combination of medications. There are no surgical options because of the body-wide distribution of the malignant cells. In general, cytotoxic chemotherapy for ALL combines multiple antileukemic drugs in various combinations. Chemotherapy for ALL consists of three phases: remission induction, intensification, and maintenance therapy.
| Phase | Description | Agents |
|---|---|---|
| Remission induction | The aim of remission induction is to rapidly kill most tumor cells and get the patient into remission. This is defined as the presence of less than 5% leukemic blasts in the bone marrow, normal blood cells and absence of tumor cells from blood, and absence of other signs and symptoms of the disease. Central nervous system (CNS) prophylaxis should begin during this phase of treatment and continue during the consolidation/intensification period. The rationale is based on the presence of CNS involvement in 10%-40% of adult patients at diagnosis. | Combination of prednisolone or dexamethasone, vincristine, asparaginase (better tolerance in pediatric patients), and daunorubicin (used in Adult ALL) is used to induce remission. Central nervous system prophylaxis can be achieved via irradiation, cytarabine + methotrexate, or liposomal cytarabine.[25] In Philadelphia chromosome-positive ALL, the intensity of initial induction treatment may be less than has been traditionally given.[26][27] |
| Consolidation/intensification | Intensification uses high doses of intravenous multidrug chemotherapy to further reduce tumor burden. Since ALL cells sometimes penetrate the CNS, most protocols include delivery of chemotherapy into the CNS fluid (termed intrathecal chemotherapy). Some centers deliver the drug through Ommaya reservoir (a device surgically placed under the scalp and used to deliver drugs to the CNS fluid and to extract CNS fluid for various tests). Other centers would perform multiple lumbar punctures as needed for testing and treatment delivery. | Typical intensification protocols use vincristine, cyclophosphamide, cytarabine, daunorubicin, etoposide, thioguanine or mercaptopurine given as blocks in different combinations. For CNS protection, intrathecal methotrexate or cytarabine is usually used combined with or without cranio-spinal irradiation (the use of radiation therapy to the head and spine). Central nervous system relapse is treated with intrathecal administration of hydrocortisone, methotrexate, and cytarabine. |
| Maintenance therapy | The aim of maintenance therapy is to kill any residual cell that was not killed by remission induction and intensification regimens. Although such cells are few, they will cause relapse if not eradicated. | For this purpose, daily oral mercaptopurine, once weekly oral methotrexate, once monthly 5-day course of intravenous vincristine and oral corticosteroids are usually used. The length of maintenance therapy is 3 years for boys, 2 years for girls and adults.[28] |
As the chemotherapy regimens can be intensive and protracted (often about 2 years in case of the GMALL UKALL, HyperCVAD, or CALGB protocols; for ALL, about 3 years, 2 months for males on COG protocols, and 2 years, 2 months for females — longer for males because the testicles are a potential reservoir), many patients have an intravenous catheter inserted into a large vein (termed a central venous catheter or a Hickman line), or a Portacath, a cone-shaped port with a silicone nose that is surgically planted under the skin, usually near the collar bone, and the most effective product available, due to low infection risks and the long-term viability of a portacath.
Radiation therapy
Radiation therapy (or radiotherapy) is used on painful bony areas, in high disease burdens, or as part of the preparations for a bone marrow transplant (total body irradiation). Radiation in the form of whole-brain radiation is also used for central nervous system prophylaxis, to prevent recurrence of leukemia in the brain. Whole-brain prophylaxis radiation used to be a common method in treatment of children’s ALL. Recent studies showed that CNS chemotherapy provided results as favorable but with less developmental side-effects. As a result, the use of whole-brain radiation has been more limited. Most specialists in adult leukemia have abandoned the use of radiation therapy for CNS prophylaxis, instead using intrathecal chemotherapy.
Biological therapy
For some subtypes of relapsed ALL, aiming at biological targets such as the proteasome, in combination with chemotherapy, has given promising results in clinical trials.[29] Selection of biological targets on the basis of their combinatorial effects on the leukemic lymphoblasts can lead to clinical trials for improvement in the effects of ALL treatment.[30]
In ongoing clinical trials, a CD19-CD3 bi-specific monoclonal murine antibody, blinatumomab, is showing great promise.
Immunotherapy
Chimeric antigen receptors (CARs) have been developed as a promising therapy for ALL. This technology uses a single chain variable fragment (scFv) designed to recognize the cell surface marker CD19 as a method of treating ALL. CD19 is a molecule found on all B-cells and can be used as a means of distinguishing the potentially malignant B-cell population in the patient. In this therapy, mice are immunized with the CD19 antigen and produce anti-CD19 antibodies. Hybridomas developed from the mouse spleen cells fused to a myeloma cell line can be developed as a source for the cDNA encoding the CD19 specific antibody.[31] The cDNA is sequenced and the sequence encoding the variable heavy and variable light chains of these antibodies are cloned together using a small peptide linker. This resulting sequence encodes the scFv. This can be cloned into a transgene encoding what will become the endodomain of the CAR. There are varying arrangements of subunits used as the endodomain but they generally consist of the hinge region that attaches to the scFv, a transmembrane region, the intracellular region of a costimulatory molecule such as CD28, and the intracellular domain of CD3-zeta containing ITAM repeats. Other sequences frequently included are: 4-1bb and OX40.[32] The final transgene sequence, containing the scFv and endodomain sequences is then inserted into immune effector cells that are obtained from the patient and expanded in vitro. In previous trials these have been a type of T-cell capable of cytotoxicity.[33] Inserting the DNA into the effector cell can be accomplished by several methods. Most commonly, this is done using a lentivirus which encodes the transgene. Pseudotyped, self-inactivating lentiviruses have been shown to be an effective method for the stable insertion of a desired transgene into the target cell genomic DNA.[34] Other methods include electroporation and transfection but these are limited in their efficacy as transgene expression will diminish over time. The gene-modified effector cells are then transplanted back into the patient. Typically this process is done in conjunction with a conditioning regimen such as cyclophosphamide which has been shown to potentiate the effects of infused T-cells. This effect has been attributed to making an immunologic space within which the cells populate.[32] The process as a whole results in an effector cell, typically a T-cell, that can recognize a tumor cell antigen in a manner that is independent of the major histocompatibility complex, and which can initiate a cytotoxic response.
Pregnancy
Leukemia is rarely associated with pregnancy, affecting only about 1 in 10,000 pregnant women.[35] How it is handled depends primarily on the type of leukemia. Acute leukemias normally require prompt, aggressive treatment, despite significant risks of pregnancy loss and birth defects, especially if chemotherapy is given during the developmentally sensitive first trimester.[35]
It is possible, although extremely rare, for leukemia to spread from the mother to the child.[36] This is called vertical transmission.
Prognosis
The 5-year survival rate for children who have ALL has improved from zero six decades ago, to 85% currently, largely because of clinical trials on new chemotherapeutic agents and improvements in stem cell transplantation (SCT) technology.[37]
Five-year survival rates evaluate older, not current, treatments. New drugs, and matching treatment to the genetic characteristics of the blast cells, may improve those rates. The prognosis for ALL differs among individuals depending on a variety of factors:
- Gender: Females tend to fare better than males.
- Ethnicity: Caucasians are more likely to develop acute leukemia than African-Americans, Asians, or Hispanics. However, they also tend to have a better prognosis than non-Caucasians.
- Age at diagnosis: children 1–10 years of age are most likely to develop ALL and to be cured of it. Cases in older patients are more likely to result from chromosomal abnormalities (e.g., the Philadelphia chromosome) that make treatment more difficult and prognoses poorer.
- White blood cell count at diagnosis of less than 50,000/µl
- Cancer spread into the Central nervous system (brain or spinal cord) has worse outcomes.
- Morphological, immunological, and genetic subtypes
- Patient's response to initial treatment
- Genetic disorders such as Down syndrome
Cytogenetics, the study of characteristic large changes in the chromosomes of cancer cells, is an important predictor of outcome.[38]
Some cytogenetic subtypes have a worse prognosis than others. These include:[1]
- A translocation between chromosomes 9 and 22, known as the Philadelphia chromosome, occurs in about 20% of adult and 5% in pediatric cases of ALL.
- A translocation between chromosomes 4 and 11 occurs in about 4% of cases and is most common in infants under 12 months.
- Not all translocations of chromosomes carry a poorer prognosis. Some translocations are relatively favorable. For example, hyperdiploidy (>50 chromosomes) is a good prognostic factor.
- Genome-wide copy number changes can be assessed by conventional cytogenetics or virtual karyotyping. SNP array virtual karyotyping can detect copy number changes and LOH status, while arrayCGH can detect only copy number changes. Copy neutral LOH (acquired uniparental disomy) has been reported at key loci in ALL, such as CDKN2A gene, which have prognostic significance.[39][40][41] SNP arrayvirtual karyotyping can readily detect copy neutral LOH. Array CGH, FISH, and conventional cytogenetics cannot detect copy neutral LOH.
| Cytogenetic change | Risk category |
|---|---|
| Philadelphia chromosome | Poor prognosis |
| t(4;11)(q21;q23) | Poor prognosis |
| t(8;14)(q24.1;q32) | Poor prognosis |
| Complex karyotype (more than four abnormalities) | Poor prognosis |
| Low hypodiploidy or near triploidy | Poor prognosis |
| High hyperdiploidy (specifically, trisomy 4, 10, 17) | Good prognosis |
| del(9p) | Good prognosis |
Correlation of prognosis with bone marrow cytogenetic finding in acute lymphoblastic leukemia
| Prognosis | Cytogenetic findings |
|---|---|
| Favorable | Hyperdiploidy > 50 ; t (12;21) |
| Intermediate | Hyperdiploidy 47 -50; Normal(diploidy); del (6q); Rearrangements of 8q24 |
| Unfavorable | Hypodiploidy-near haploidy; Near tetraploidy; del (17p); t (9;22); t (11q23) |
Unclassified ALL is considered to have an intermediate prognosis.[42]
Epidemiology
Acute lymphoblastic leukemia is seen in both children and adults; the highest incidence is seen between ages 2 and 5 years.[1]> ALL is the most common childhood cancer,[21]:1382 constituting about 23[20]:491 to 30% of cancers before age 15.[21]:1527 Although 80 to 90% of children will have a durable complete response with treatment[21]:1527 it is the leading cause of cancer-related deaths among children.[43] In adults, ALL is less common than acute myelogenous leukemia (AML). However, as there are more adults than children, the number of cases of ALL seen in adults is comparable to that seen in children.[1]
International
Internationally, ALL is more common in children with Caucasian descent, being more common in Hispanics and in Latin America than in Africa.[3]:1617[4] Epidemiological studies suggest that environmental factors on their own make only a minor contribution to disease risk, but environmental factors may interact with genetics.[44] Genome-wide association studies have found associations with a number of genetic single-nucleotide polymorphisms, including ARID5B, IKZF1 and CEBPE.[21]:1388[43][45][46][47]
There is an increased incidence in people with Down syndrome, Fanconi anemia, Bloom syndrome, ataxia telangiectasia, X-linked agammaglobulinemia, and severe combined immunodeficiency. There is an increased risk in people with a family history of autoimmune diseases, particularly autoimmune thyroid diseases (namely Graves' disease or Hashimoto's thyroiditis).[48]
United States
In the United States, the annual incidence of ALL is roughly 6,000[2] 3,000-3,500, or approximately 1 in 50,000. ALL is slightly more common in males than females. In the United States in 2010, incidence from birth to age 19 was 38.4 per 1,000,000 per year in boys and 30.2 per 1,000,000 per year in girls.[49] Prevalence was 30,171, and observed survival was 90% (based on data from 2003-2009). ALL has a bimodal age distribution, having a high incidence in ages 2–5 and another peak in incidence above 50 years old.
United Kingdom
ALL accounts for 8% of all leukemia cases in the United Kingdom; around 650 people were diagnosed with the disease in 2011.[50]
References
- 1 2 3 4 5 6 7 8 9 10 Seiter, K (5 February 2014). Sarkodee-Adoo, C; Talavera, F; Sacher, RA; Besa, EC, eds. "Acute Lymphoblastic Leukemia". Medscape Reference. WebMD. Retrieved 17 April 2014.
- 1 2 Inaba H, Greaves M, Mullighan CG (June 2013). "Acute lymphoblastic leukaemia." (PDF). Lancet. 381 (9881): 1943–55. doi:10.1016/S0140-6736(12)62187-4. PMC 3816716
 . PMID 23523389.
. PMID 23523389. - 1 2 Greer, J. P.; Arber, D. A.; Glader, B.; et al. (2013). Wintrobe's Clinical Hematology (13th ed.). Lippincott Williams & Wilkins. ISBN 9781451172683.
- 1 2 Urayama KY, Manabe A (2014). "Genomic evaluations of childhood acute lymphoblastic leukemia susceptibility across race/ethnicities". Rinsho Ketsueki. 55 (10): 2242–8. PMID 25297793.
- ↑ "Evolution of cancer treatments: Chemotherapy" (PDF). Cancer.org. American Cancer Society. 8 June 2012.
- 1 2 Mukherjee, Siddhartha (2010). The Emperor of all Maladies: a Biography of Cancer (1st Scribner hardcover ed.). New York: Scribner. ISBN 978-1-4391-0795-9.
- ↑ "Leukemia--Acute Lymphocytic". American Cancer Society. Retrieved 8 February 2013.
- ↑ Smith MA, Rubinstein L, Anderson JR, et al. (Feb 1999). "Secondary Leukemia or Myelodysplastic Syndrome After Treatment With Epipodophyllotoxins" (PDF). Journal of Clinical Oncology. American Society for Clinical Oncology. 17 (2): 569–77. PMID 10080601.
- ↑ Collier, J.A.B (1991). Oxford Handbook of Clinical Specialties, Third Edition. Oxford. p. 810. ISBN 0-19-262116-5.
- ↑ Longo, D (2011). "Chapter 110: Malignancies of Lymphoid Cells". Harrison's Principles of Internal Medicine (18 ed.). New York: McGraw-Hill Professional. ISBN 978-0-07174889-6.
- ↑ Rytting, ME, ed. (November 2013). "Acute Leukemia". Merck Manual Professional. Merck Sharp & Dohme Corp. Retrieved 17 April 2014.
- ↑ Stams WA, den Boer ML, Beverloo HB, Meijerink JP, van Wering ER, Janka-Schaub GE, Pieters R (April 2005). "Expression levels of TEL, AML1, and the fusion products TEL-AML1 and AML1-TEL versus drug sensitivity and clinical outcome in t(12;21)-positive pediatric acute lymphoblastic leukemia". Clin. Cancer Res. 11 (8): 2974–80. doi:10.1158/1078-0432.CCR-04-1829. PMID 15837750.
- 1 2 3 4 Pakakasama S, Kajanachumpol S, Kanjanapongkul S, et al. (August 2008). "Simple multiplex RT-PCR for identifying common fusion transcripts in childhood acute leukemia". Int J Lab Hematol. 30 (4): 286–91. doi:10.1111/j.1751-553X.2007.00954.x. PMID 18665825.
- ↑ McWhirter JR, Neuteboom ST, Wancewicz EV, et al. (September 1999). "Oncogenic homeodomain transcription factor E2A-Pbx1 activates a novel WNT gene in pre-B acute lymphoblastoid leukemia". Proc. Natl. Acad. Sci. U.S.A. 96 (20): 11464–9. doi:10.1073/pnas.96.20.11464. PMC 18056. PMID 10500199.
- ↑ Rudolph C, Hegazy AN, von Neuhoff N, et al. (August 2005). "Cytogenetic characterization of a BCR-ABL transduced mouse cell line". Cancer Genet. Cytogenet. 161 (1): 51–6. doi:10.1016/j.cancergencyto.2004.12.021. PMID 16080957.
- ↑ Caslini C, Serna A, Rossi V, et al. (June 2004). "Modulation of cell cycle by graded expression of MLL-AF4 fusion oncoprotein". Leukemia. 18 (6): 1064–71. doi:10.1038/sj.leu.2403321. PMID 14990976.
- ↑ Martín-Subero JI, Odero MD, Hernandez R, Cigudosa JC, Agirre X, Saez B, Sanz-García E, Ardanaz MT, Novo FJ, Gascoyne RD, Calasanz MJ, Siebert R (August 2005). "Amplification of IGH/MYC fusion in clinically aggressive IGH/BCL2-positive germinal center B-cell lymphomas". Genes Chromosomes Cancer. 43 (4): 414–23. doi:10.1002/gcc.20187. PMID 15852472.
- ↑ Zalcberg IQ, Silva ML, Abdelhay E, et al. (October 1995). "Translocation 11;14 in three children with acute lymphoblastic leukemia of T-cell origin". Cancer Genet. Cytogenet. 84 (1): 32–8. doi:10.1016/0165-4608(95)00062-3. PMID 7497440.
- ↑ "ACS :: How Is Acute Lymphocytic Leukemia Classified?".
- 1 2 3 DeAngelo DJ, Pui C. Acute lymphoblastic leukemia and lymphoblastic lymphoma. Chapter 19 of American Society of Hematology Self-Assessment Program. 2013. ISBN 9780982843512
- 1 2 3 4 5 6 Orkin, S. H.; Nathan, D. G.; Ginsburg, D.; et al. (2014). Nathan and Oski's Hematology and Oncology of Infancy and Childhood (8th ed.). Saunders. ISBN 9781455754144.
- ↑ "Advances in Acute Lymphoblastic Leukemia | Clinical Laboratory Science | Find Articles at BNET.com". Clinical Laboratory Science. 2004.
- ↑ Seiter K, Harris JE. Acute Lymphoblastic Leukemia Staging
- ↑ Acute lymphoblastic leukemia at Mount Sinai Hospital
- ↑ Jabbour E, Thomas D, Cortes J, et al. (May 15, 2010). "Central nervous system prophylaxis in adults with acute lymphoblastic leukemia: current and emerging therapies.". Cancer. 116 (10): 2290–300. doi:10.1002/cncr.25008. PMID 20209620.
- ↑ Yanada M (2015). "Time to tune the treatment of Ph+ ALL". Blood. 125 (24): 3674–5. doi:10.1182/blood-2015-04-641704. PMID 26069331.
- ↑ Seiter K, Harris JE. Acute Lymphoblastic Leukemia Treatment Protocols. emedicine; Medscape.
- ↑ Hoffbrand, Victor; Moss, Paul; Pettit, John (31 October 2006). Essential Haematology. Wiley. ISBN 978-1-4051-3649-5. Retrieved 14 September 2013.
- ↑ Messinger YH, Gaynon PS, Sposto R, et al. (July 2012). "Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study". Blood. 120 (2): 285–90. doi:10.1182/blood-2012-04-418640. PMID 22653976.
- ↑ Lambrou GI, Papadimitriou L, Chrousos GP, Vlahopoulos SA (January 2012). "Glucocorticoid and proteasome inhibitor impact on the leukemic lymphoblast: multiple, diverse signals converging on a few key downstream regulators". Mol Cell Endocrinol. 351 (2): 142–51. doi:10.1016/j.mce.2012.01.003. PMID 22273806.
- ↑ Grupp SA, Kalos M, Barrett D, et al. (2013). "Chimeric antigen receptor-modified T cells for acute lymphoid leukemia". N. Engl. J. Med. 368 (16): 1509–18. doi:10.1056/NEJMoa1215134. PMC 4058440. PMID 23527958.
- 1 2 Barrett DM, Singh N, Porter DL, et al. (2014). "Chimeric antigen receptor therapy for cancer". Annu. Rev. Med. 65: 333–47. doi:10.1146/annurev-med-060512-150254. PMC 4120077. PMID 24274181.
- ↑ Alonso-Camino V, Sánchez-Martín D, Compte M, et al. (2013). "CARbodies: Human Antibodies Against Cell Surface Tumor Antigens Selected From Repertoires Displayed on T Cell Chimeric Antigen Receptors". Mol Ther Nucleic Acids. 2: e93. doi:10.1038/mtna.2013.19. PMID 23695536.
- ↑ Zufferey R, Dull T, Mandel RJ, et al. (1998). "Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery". J. Virol. 72 (12): 9873–80. PMC 110499. PMID 9811723.
- 1 2 Shapira T, Pereg D, Lishner M (September 2008). "How I treat acute and chronic leukemia in pregnancy". Blood Rev. 22 (5): 247–59. doi:10.1016/j.blre.2008.03.006. PMID 18472198.
- ↑ Isoda T, Ford AM, Tomizawa D, et al. (October 2009). "Immunologically silent cancer clone transmission from mother to offspring". Proc. Natl. Acad. Sci. U.S.A. 106 (42): 17882–5. doi:10.1073/pnas.0904658106. PMC 2764945. PMID 19822752.
- ↑ Park KD (March 2014). "How do we prepare ourselves for a new paradigm of medicine to advance the treatment of pediatric acute lymphoblastic leukemia?". Blood Research. 49 (1): 3–4. doi:10.5045/br.2014.49.1.3. PMC 3974954. PMID 24724058.
- ↑ Moorman AV, Harrison CJ, Buck GA, et al. (Apr 15, 2007). "Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial.". Blood. 109 (8): 3189–3197. doi:10.1182/blood-2006-10-051912. PMID 17170120.
- ↑ Kawamata N, Ogawa S, Zimmermann M, Kato M, Sanada M, Hemminki K, Yamatomo G, Nannya Y, Koehler R, Flohr T, Miller CW, Harbott J, Ludwig WD, Stanulla M, Schrappe M, Bartram CR, Koeffler HP (Jan 15, 2008). "Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray.". Blood. 111 (2): 776–784. doi:10.1182/blood-2007-05-088310. PMC 2200831. PMID 17890455.
- ↑ Bungaro S, Dell'Orto MC, Zangrando A, et al. (1 January 2009). "Integration of genomic and gene expression data of childhood ALL without known aberrations identifies subgroups with specific genetic hallmarks". Genes, Chromosomes and Cancer. 48 (1): 22–38. doi:10.1002/gcc.20616. PMID 18803328.
- ↑ Sulong S, Moorman AV, Irving JA, et al. (Jan 1, 2009). "A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups.". Blood. 113 (1): 100–107. doi:10.1182/blood-2008-07-166801. PMID 18838613.
- ↑ Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. (January 2009). "A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study". Lancet Oncol. 10 (2): 125–34. doi:10.1016/S1470-2045(08)70339-5. PMC 2707020. PMID 19138562.
- 1 2 Guo LM, Xi JS, Ma Y, et al. (2014). "ARID5B gene rs10821936 polymorphism is associated with childhood acute lymphoblastic leukemia: a meta-analysis based on 39,116 subjects". Tumour Biol. 35 (1): 709–13. doi:10.1007/s13277-013-1097-0. PMID 23975371.
- ↑ Evans TJ, Milne E, Anderson D, et al. (2014). "Confirmation of childhood acute lymphoblastic leukemia variants, ARID5B and IKZF1, and interaction with parental environmental exposures". PLoS ONE. 9 (10): e110255. doi:10.1371/journal.pone.0110255. PMC 4195717. PMID 25310577.
- ↑ Brisson GD, Alves LR, Pombo-de-Oliveira MS (2015). "Genetic susceptibility in childhood acute leukaemias: a systematic review". Ecancermedicalscience. 9: 539. doi:10.3332/ecancer.2015.539. PMC 4448992. PMID 26045716.
- ↑ Rudant J, Orsi L, Bonaventure A, et al. (2015). "ARID5B, IKZF1 and non-genetic factors in the etiology of childhood acute lymphoblastic leukemia: the ESCALE study". PLoS ONE. 10 (3): e0121348. doi:10.1371/journal.pone.0121348. PMC 4373901. PMID 25806972.
- ↑ Hsu LI, Chokkalingam AP, Briggs FB, et al. (2015). "Association of genetic variation in IKZF1, ARID5B, and CEBPE and surrogates for early-life infections with the risk of acute lymphoblastic leukemia in Hispanic children". Cancer Causes Control. 26 (4): 609–19. doi:10.1007/s10552-015-0550-3. PMID 25761407.
- ↑ Perillat-Menegaux F, Clavel J, Auclerc MF, et al. (2003). "Family History of Autoimmune Thyroid Disease and Childhood Acute Leukemia". Cancer Epidemiology, Biomarkers & Prevention. 12 (1): 60–3. PMID 12540505. Retrieved 11 April 2014.
A statistically significant association between a history of autoimmune disease in first- or second-degree relatives and ALL (OR, 1.7; 95% confidence interval (CI), 1.0–2.8) was found. A relationship between thyroid diseases overall and ALL...was observed. This association was more pronounced for potentially autoimmune thyroid diseases (Grave’s disease and/or hyperthyroidism and Hashimoto’s disease and/or hypothyroidism) (OR, 3.5; 95% CI, 1.1–10.7 and OR, 5.6; 95% CI, 1.0–31.1, respectively for ALL and ANLL), whereas it was not statistically significant for the other thyroid diseases...The results suggest that a familial history of autoimmune thyroid disease may be associated with childhood acute leukemia.
- ↑ Ward E, DeSantis C, Robbins A, Kohler B, Jemal A (Mar–Apr 2014). "Childhood and adolescent cancer statistics, 2014.". CA: A Cancer Journal for Clinicians. 64 (2): 83–103. doi:10.3322/caac.21219. PMID 24488779.
- ↑ "Acute lymphoblastic leukaemia (ALL) statistics". Cancer Research UK. Retrieved 27 October 2014.